 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2xwc: Crystal structure of the DNA binding domain of human TP73 refined... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xwc | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the DNA binding domain of human TP73 refined at 1.8 A resolution | |||||||||
 Components Components | TUMOUR PROTEIN P73 | |||||||||
 Keywords Keywords | DNA BINDING PROTEIN / DNA-BINDING PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of lung ciliated cell differentiation / negative regulation of cardiac muscle cell proliferation / TP53 Regulates Transcription of Death Receptors and Ligands / Activation of PUMA and translocation to mitochondria / Regulation of TP53 Activity through Association with Co-factors / TP53 Regulates Transcription of Caspase Activators and Caspases / TP53 Regulates Transcription of Genes Involved in Cytochrome C Release / TP53 regulates transcription of several additional cell death genes whose specific roles in p53-dependent apoptosis remain uncertain / intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator / mismatch repair ...positive regulation of lung ciliated cell differentiation / negative regulation of cardiac muscle cell proliferation / TP53 Regulates Transcription of Death Receptors and Ligands / Activation of PUMA and translocation to mitochondria / Regulation of TP53 Activity through Association with Co-factors / TP53 Regulates Transcription of Caspase Activators and Caspases / TP53 Regulates Transcription of Genes Involved in Cytochrome C Release / TP53 regulates transcription of several additional cell death genes whose specific roles in p53-dependent apoptosis remain uncertain / intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator / mismatch repair / regulation of mitotic cell cycle / MDM2/MDM4 family protein binding / transcription corepressor binding / protein tetramerization / promoter-specific chromatin binding / intrinsic apoptotic signaling pathway in response to DNA damage / p53 binding / RUNX1 regulates transcription of genes involved in differentiation of HSCs / regulation of gene expression / DNA-binding transcription activator activity, RNA polymerase II-specific / regulation of apoptotic process / DNA-binding transcription factor binding / RNA polymerase II-specific DNA-binding transcription factor binding / DNA-binding transcription factor activity, RNA polymerase II-specific / regulation of cell cycle / transcription cis-regulatory region binding / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of cell population proliferation / DNA damage response / protein kinase binding / positive regulation of DNA-templated transcription / chromatin / DNA-templated transcription / positive regulation of transcription by RNA polymerase II / nucleoplasm / metal ion binding / identical protein binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.82 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.82 Å | |||||||||
 Authors Authors | Canning, P. / Zhang, Y. / Vollmar, M. / Krojer, T. / Ugochukwu, E. / Muniz, J.R.C. / von Delft, F. / Weigelt, J. / Arrowsmith, C.H. / Edwards, A.M. ...Canning, P. / Zhang, Y. / Vollmar, M. / Krojer, T. / Ugochukwu, E. / Muniz, J.R.C. / von Delft, F. / Weigelt, J. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A.N. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2012 Title: Structural Basis for Aspp2 Recognition by the Tumor Suppressor P73. Authors: Canning, P. / von Delft, F. / Bullock, A.N. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xwc.cif.gz | 98.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xwc.ent.gz | 75.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2xwc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xw/2xwcftp://data.pdbj.org/pub/pdb/validation_reports/xw/2xwc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4a63C  20cjS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23342.561 Da / Num. of mol.: 1 / Fragment: DNA-BINDING DOMAIN, RESIDUES 112-311 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PNIC-CTHF / Production host:  | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-TAM / | 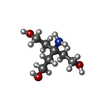  Mass: 163.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM Mass: 163.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 175 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 175 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Has protein modification | Y | Nonpolymer details | IDENTITY OF 2 ZN IONS CONFIRMED BY MAD EXPERIMENT. PEAKS IDENTIFIED AT ZN BINDING SITES IN ...IDENTITY OF 2 ZN IONS CONFIRMED BY MAD EXPERIMENT | Sequence details | RESIDUES 312-318 ARE DUE TO CLONING. INITIATOR METHIONINE | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.13 % / Description: NONE |
|---|---|
| Crystal grow | Details: 1.2M SODIUM POTASSIUM TARTRATE, 0.25% PEG MME 5000, 0.1M TRIS PH 9 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.9795 / Beamline: I02 / Wavelength: 0.9795 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: May 26, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 1.82→49.36 Å / Num. obs: 280763 / % possible obs: 100 % / Observed criterion σ(I): 2 / Redundancy: 13.2 % / Rmerge(I) obs: 0.16 / Net I/σ(I): 11.9 |
| Reflection shell | Resolution: 1.82→49.36 Å / Redundancy: 12.9 % / Rmerge(I) obs: 1.24 / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 20CJ Resolution: 1.82→49.36 Å / Cor.coef. Fo:Fc: 0.951 / Cor.coef. Fo:Fc free: 0.93 / SU B: 4.788 / SU ML: 0.081 / Cross valid method: THROUGHOUT / ESU R: 0.13 / ESU R Free: 0.126 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. INITIATOR MET COULD NOT BE RESOLVED OWING TO DISORDER.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.202 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.82→49.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|