 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2xtv: Structure of E.coli rhomboid protease GlpG, active site mutant, S... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xtv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of E.coli rhomboid protease GlpG, active site mutant, S201T, orthorhombic crystal form | ||||||
 Components Components | RHOMBOID PROTEASE GLPG | ||||||
 Keywords Keywords | HYDROLASE / MEMBRANE PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationrhomboid protease / endopeptidase activity / serine-type endopeptidase activity / proteolysis / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Vinothkumar, K.R. | ||||||
 Citation Citation | Journal: J. Mol. Biol. / Year: 2011 Title: Structure of rhomboid protease in a lipid environment. Authors: Vinothkumar, K.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xtv.cif.gz | 62 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xtv.ent.gz | 42.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2xtv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xt/2xtvftp://data.pdbj.org/pub/pdb/validation_reports/xt/2xtv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2xtuC  2xovS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 20256.033 Da / Num. of mol.: 1 / Fragment: CORE TM DOMAIN, RESIDUES 93-272 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MC3 / 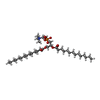  Mass: 677.933 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C36H72NO8P / Comment: phospholipid*YM Mass: 677.933 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C36H72NO8P / Comment: phospholipid*YM#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 79 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 79 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Nonpolymer details | IN THE PRESENT MODEL LIPIDS (MC3) MOLECULES 504, 505, 508, 511, 513 AND 514 COULD ALSO REPRESENT ...IN THE PRESENT MODEL LIPIDS (MC3) MOLECULES 504, 505, 508, 511, 513 AND 514 COULD ALSO REPRESENT DETERGENT NONYL GLUCOSIDE USED FOR PURIFICATI | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 48.94 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 1.5M NACL, 0.1M BIS-TRIS, PH7, 2% DMPC/CHAPSO, (ADDED ONLY TO THE PROTEIN), 298K, pH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 / Beamline: I03 / Wavelength: 0.9763 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Apr 29, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→45.54 Å / Num. obs: 22469 / % possible obs: 95.8 % / Observed criterion σ(I): 0 / Redundancy: 3.3 % / Biso Wilson estimate: 20.3 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 13.3 |
| Reflection shell | Resolution: 1.7→1.79 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.31 / Mean I/σ(I) obs: 3.2 / % possible all: 86.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2XOV Resolution: 1.7→28.009 Å / SU ML: 0.19 / σ(F): 0 / Phase error: 18.38 / Stereochemistry target values: ML Details: RESIDUES 246 AND 247 ARE DISORDERED. HENCE THERE IS A GAP BETWEEN 245 AND 248.THERE IS A DENSITY ABOVE THE ACTIVE SITE WHICH HAS NOT BEEN MODELLED, AS NONE OF COMPONENTS OF CRYSTALLISATION ...Details: RESIDUES 246 AND 247 ARE DISORDERED. HENCE THERE IS A GAP BETWEEN 245 AND 248.THERE IS A DENSITY ABOVE THE ACTIVE SITE WHICH HAS NOT BEEN MODELLED, AS NONE OF COMPONENTS OF CRYSTALLISATION CAN EXPLAIN THE DENSITY.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 66.136 Å2 / ksol: 0.412 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.1 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→28.009 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
