Mass: 18.015 Da / Num. of mol.: 721 / Source method: isolated from a natural source / Formula: H2O
-
Details
Compound details
ANTIPAIN HYDROCHLORIDE IS A REVERSIBLE INHIBITOR OF SERINE/CYSTEINE PROTEASES AND SOME TRYPSIN-LIKE ...ANTIPAIN HYDROCHLORIDE IS A REVERSIBLE INHIBITOR OF SERINE/CYSTEINE PROTEASES AND SOME TRYPSIN-LIKE SERINE PROTEASES. ITS ACTION RESEMBLES LEUPEPTIN; HOWEVER, ITS PLASMIN INHIBITION IS LESS AND ITS CATHEPSIN A INHIBITION IS MORE THAN THAT OBSERVED WITH LEUPEPTIN. IT IS AN INHIBITOR OF TRYPSIN-LIKE PROTEASES (E.G. TRYPSIN, PAPAIN, CATHEPSIN B); CATHEPSIN A, A CARBOXYPEPTIDASE IS INHIBITED TOO, WHICH IS NOT TRUE FOR OTHER INHIBITORS FROM ACTINOMYCES.THE NAME IS DERIVED FROM ANTI-PAPAIN. GROUP: 1 NAME: ANTIPAIN CHAIN: B COMPONENT_1: PEPTIDE LIKE SEQUENCE RESIDUES 1 TO 4 DESCRIPTION: ANTIPAIN FORMS A COVALENT LINKAGE BETWEEN THE ARGINAL RESIDUE OF THE INHIBITOR AND SER RESIDUE OF THE PROTEIN.THE PROTEASE INHIBITOR ANTIPAIN INCREASES THE EFFECTIVENESSOF UV IRRADIATION ON CESSATION OF RESPIRATION AND CELL KILLING IN ESCHERICHIA COLI B/R CULTURES WITHOUT AFFECTING EXCISION OF PYRIMIDINE DIMERS. THE ACTIONS ARE SIMILAR TO THOSE CAUSED BY CYCLIC AMP IN IRRADIATED CULTURES. ENGINEERED RESIDUE IN CHAIN A, PHE 25 TO LEU
Has protein modification
Y
Nonpolymer details
CHLORINE ION (CL): NA
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 4.2 Å3/Da / Density % sol: 71 % / Description: NONE
Crystal grow
pH: 4.2 Details: PROTEIN WAS DIALYZED INTO 50 MM TRIS, PH 8.0 AND INCUBATED WITH 10 MM ANTIPAIN FOR 30 MINS. THIS WAS MIXED IN A 1:1 RATIO WITH 25% 1,2 PROPANEDIOL, 10% GLYCEROL, 5 % PEG300 AND 0.1 M PHOSPHATE CITRATE, PH 4.2
-
Data collection
Diffraction
ID
Mean temperature (K)
Crystal-ID
1
103
1
2
103
1
Diffraction source
Source
Site
Beamline
Type
ID
Wavelength
ROTATING ANODE
RIGAKU MICROMAX-007
1
1.541
SYNCHROTRON
ESRF
BM14
2
1.005
Detector
Type
ID
Detector
Date
Details (eV)
MARRESEARCH
1
IMAGE PLATE
Jun 10, 2008
MIRRORS
MARRESEARCH
2
CCD
Radiation
ID
Monochromator
Protocol
Monochromatic (M) / Laue (L)
Scattering type
Wavelength-ID
1
GRAPHITE
SINGLEWAVELENGTH
M
x-ray
1
2
M
x-ray
1
Radiation wavelength
ID
Wavelength (Å)
Relative weight
1
1.541
1
2
1.005
1
Reflection
Resolution: 1.65→32 Å / Num. obs: 167772 / % possible obs: 98.4 % / Observed criterion σ(I): 1.5 / Redundancy: 3.3 % / Biso Wilson estimate: 29.5 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 8.8
Reflection shell
Resolution: 1.65→1.71 Å / Redundancy: 3 % / Rmerge(I) obs: 0.46 / Mean I/σ(I) obs: 1.7 / % possible all: 100
-
Processing
Software
Name
Version
Classification
REFMAC
5.5.0102
refinement
d*TREK
datareduction
SCALA
datascaling
autoSHARP
phasing
SHELXCD
phasing
SHELXD
phasing
Refinement
Method to determine structure: SAD Starting model: NONE Resolution: 1.65→117.851 Å / Cor.coef. Fo:Fc: 0.981 / Cor.coef. Fo:Fc free: 0.971 / SU B: 3.558 / SU ML: 0.051 / Cross valid method: THROUGHOUT / ESU R: 0.066 / ESU R Free: 0.064 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES 1-9 AND 731 ARE DISORDERED.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.178
8321
5.08 %
RANDOM
Rwork
0.1403
-
-
-
obs
0.142
167169
98.007 %
-
Solvent computation
Ion probe radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL PLUS MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information LEISHMANIA MAJOR (eukaryote)
LEISHMANIA MAJOR (eukaryote) ACTINOBACTERIA (actinobacteria)
ACTINOBACTERIA (actinobacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj
 Assembly
Assembly

 Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 606.717 Da / Num. of mol.: 1 / Source method: isolated from a natural source
Type: Oligopeptide / Class: Enzyme inhibitor / Mass: 606.717 Da / Num. of mol.: 1 / Source method: isolated from a natural source
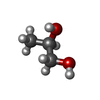
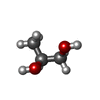




 Mass: 92.094 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 76.094 Da / Num. of mol.: 48 / Source method: obtained synthetically / Formula: C3H8O2
Mass: 76.094 Da / Num. of mol.: 48 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2
Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4 Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Sample preparation
Sample preparation
 Processing
Processing