SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
Mass: 18.015 Da / Num. of mol.: 786 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
Sequence details
THE ORF WAS IDENTIFIED FROM A PUTATIVE TRYPANOTHIONE REDUCTASE FROM T. BRUCEI STRAIN S427 IN THE ...THE ORF WAS IDENTIFIED FROM A PUTATIVE TRYPANOTHIONE REDUCTASE FROM T. BRUCEI STRAIN S427 IN THE GENEDB. THE SAME ORF OUT GB TB10.406.0520 ENTRY (FROM STRAIN S927) WAS CLONED OF A DIFFERENT T. BRUCEI BRUCEI STRAIN (STRAIN S427). THE ORF OF THE PROTEIN IS NEARLY IDENTICAL TO THE TB10.406.0520 ORF (THERE ARE ONLY 2 NUCLEOTIDES THAT ARE DIFFERENT OUT OF 1479 BASE PAIRS AND NEITHER OF THESE CHANGE THE PROTEIN SEQUENCE, WHICH IS 100% IDENTICAL BETWEEN BOTH STRAINS: S927 AND S427
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.2 Å3/Da / Density % sol: 62 % / Description: NONE
Resolution: 2.3→102.6 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.907 / SU B: 5.072 / SU ML: 0.127 / Cross valid method: THROUGHOUT / ESU R: 0.273 / ESU R Free: 0.213 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22715
2952
5.1 %
RANDOM
Rwork
0.1808
-
-
-
obs
0.18315
55398
94.81 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

















 PDBj
PDBj




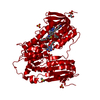
 Assembly
Assembly



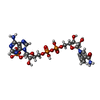



 Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM
Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3
Mass: 745.421 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM
Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Sample preparation
Sample preparation Processing
Processing