 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2wa7: Structure of the M202V mutant of human filamin b actin binding do... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2wa7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the M202V mutant of human filamin b actin binding domain at 1.85 Angstrom resolution | ||||||
 Components Components | FILAMIN-B | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / DISEASE MUTATION / SKELETAL DYSPLASIA / ACTIN-CROSSLINKING / MYOGENESIS / CYTOSKELETON / ACTIN-BINDING / CALPONIN HOMOLOGY DOMAIN / PHOSPHOPROTEIN / DIFFERENTIATION / ATELOSTEOGENESIS / MUTANT / FILAMIN / DWARFISM / CH DOMAIN / DEVELOPMENTAL PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationmuscle organ development / brush border / phagocytic vesicle / stress fiber / ISG15 antiviral mechanism / Z disc / actin filament binding / actin cytoskeleton / actin cytoskeleton organization / actin binding ...muscle organ development / brush border / phagocytic vesicle / stress fiber / ISG15 antiviral mechanism / Z disc / actin filament binding / actin cytoskeleton / actin cytoskeleton organization / actin binding / cell cortex / cell differentiation / cadherin binding / focal adhesion / neuronal cell body / signal transduction / RNA binding / extracellular exosome / membrane / identical protein binding / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Sawyer, G.M. / Clark, A.R. / Robertson, S.P. / Sutherland-Smith, A.J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2009 Title: Disease-Associated Substitutions in the Filamin B Actin Binding Domain Confer Enhanced Actin Binding Affinity in the Absence of Major Structural Disturbance: Insights from the Crystal ...Title: Disease-Associated Substitutions in the Filamin B Actin Binding Domain Confer Enhanced Actin Binding Affinity in the Absence of Major Structural Disturbance: Insights from the Crystal Structures of Filamin B Actin Binding Domains. Authors: Sawyer, G.M. / Clark, A.R. / Robertson, S.P. / Sutherland-Smith, A.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2wa7.cif.gz | 66.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2wa7.ent.gz | 47.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2wa7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wa/2wa7ftp://data.pdbj.org/pub/pdb/validation_reports/wa/2wa7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2wa5SC  2wa6C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







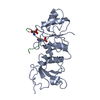








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28103.121 Da / Num. of mol.: 1 / Fragment: ACTIN-BINDING DOMAIN, RESIDUES 2-242 / Mutation: YES Source method: isolated from a genetically manipulated source Details: ARSENIC SULFUR LINK AT CYS 178 / Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PPROEXHTB-FLNB-ABDM202V / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CAC / 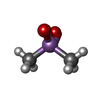  Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2 | ||||||||
| #3: Chemical |   Mass: 60.009 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CO3 Mass: 60.009 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 204 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Nonpolymer details | CACODYLATE | Sequence details | THE SEQUENCE IS DESCRIBED IN KRAKOW ET AL. (2004) NAT GENET, 36 (4) PGS 405-410. CLEAVAGE WITH RTEV ...THE SEQUENCE IS DESCRIBED IN KRAKOW ET AL. (2004) NAT GENET, 36 (4) PGS 405-410. CLEAVAGE WITH RTEV PROTEASE LEAVES THE LEADER GLY ALA MET ALA STARTING AT -2. CLONING INTO NCOI CREATED MET ALA INSTEAD OF MET AT POSITION 1. | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 45 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN WAS CRYSTALLIZED FROM 20% PEG 8000, 0.2 M MAGNESIUM ACETATE AND 0.1 M SODIUM CACODYLATE PH 6.5. |
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Mar 22, 2007 / Details: OSMIC BLUE CONFOCAL MIRRORS |
| Radiation | Monochromator: OSMIC MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→27.83 Å / Num. obs: 23537 / % possible obs: 95.5 % / Observed criterion σ(I): 2.2 / Redundancy: 4.7 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 25.8 |
| Reflection shell | Resolution: 1.85→1.95 Å / Redundancy: 4.1 % / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 2.2 / % possible all: 79.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2WA5 Resolution: 1.85→27.83 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.94 / SU B: 6.102 / SU ML: 0.098 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.141 / ESU R Free: 0.136 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES -2-11, 133-136, 241-242 ARE DISORDERED. RESIDUES GLY ALA MET ALA PRO VAL THR GLU LYS ASP LEU ALA GLU ASP (-2- 11), ASP ASP ASP ALA ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES -2-11, 133-136, 241-242 ARE DISORDERED. RESIDUES GLY ALA MET ALA PRO VAL THR GLU LYS ASP LEU ALA GLU ASP (-2- 11), ASP ASP ASP ALA (133-136), AND LYS LEU ( 241-242) WERE EXCLUDED DUE TO INSUFFICIENT ELECTRON DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.187 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→27.83 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|