 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2w3v: MYCOBACTERIUM AVIUM DIHYDROFOLATE REDUCTASE COMPLEXED WITH NADPH ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2w3v | ||||||
|---|---|---|---|---|---|---|---|
| Title | MYCOBACTERIUM AVIUM DIHYDROFOLATE REDUCTASE COMPLEXED WITH NADPH AND TRIMETHOPRIM | ||||||
 Components Components | DIHYDROFOLATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / NONCLASSICAL ANTIFOLATES / ONE-CARBON METABOLISM / LIPOPHILIC ANTIFOLATES / NADP / REDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationNADP+ binding / dihydrofolate metabolic process / dihydrofolate reductase / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / one-carbon metabolic process / cytosol Similarity search - Function | ||||||
| Biological species |  Mycobacterium avium (bacteria) Mycobacterium avium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.89 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.89 Å | ||||||
 Authors Authors | Leung, A.K.W. / Reynolds, R.C. / Borhani, D.W. | ||||||
 Citation Citation | Journal: To be Published Title: Structural Basis for Selective Inhibition of Mycobacterium Avium Dihydrofolate Reductase by a Lipophilic Antifolate Authors: Leung, A.K.W. / Ross, L.J. / Zywno-Van Ginkel, S. / Reynolds, R.C. / Seitz, L.E. / Pathak, V. / Barrow, W.W. / White, E.L. / Suling, W.J. / Piper, J.R. / Borhani, D.W. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2w3v.cif.gz | 54.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2w3v.ent.gz | 37.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2w3v.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w3/2w3vftp://data.pdbj.org/pub/pdb/validation_reports/w3/2w3v | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2w3aC  2w3bC  2w3mC  2w3wC  1rx1S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 18451.822 Da / Num. of mol.: 1 / Fragment: RESIDUES 1-167 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium avium (bacteria)Description: STRAIN OBTAINED FROM THE AIDS RESEARCH AND REFERENCE REAGENT PROGRAM, DIVISION OF AIDS, NIAID, NIH, CATALOG NUMBER 1786. Plasmid: PET11A-MACDHFR-DEL / Production host: |
|---|---|
| #2: Chemical | ChemComp-NDP / 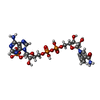  Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 |
| #3: Chemical | ChemComp-TOP / 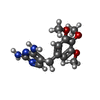  Mass: 290.318 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H18N4O3 / Comment: antibiotic*YM Mass: 290.318 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H18N4O3 / Comment: antibiotic*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | RESIDUES 168-181 OF WILDTYPE M. AVIUM DHFR WERE DELETED IN THE EXPRESSION CONSTRUCT. THE FIRST ...RESIDUES 168-181 OF WILDTYPE M. AVIUM DHFR WERE DELETED IN THE EXPRESSION |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.9 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: MAVDHFR (15 MG/ML) IN 20 MM HEPES (PH 7.0), 1 MM DTT, 0.1 MM EDTA, 1.5 MM NAN3 WAS MIXED WITH NADPH (3 MM, 15 MIN AT ROOM TEMPERATURE) AND THEN TRIMETHOPRIM (2 MM, 15 MIN ON ICE). ...Details: MAVDHFR (15 MG/ML) IN 20 MM HEPES (PH 7.0), 1 MM DTT, 0.1 MM EDTA, 1.5 MM NAN3 WAS MIXED WITH NADPH (3 MM, 15 MIN AT ROOM TEMPERATURE) AND THEN TRIMETHOPRIM (2 MM, 15 MIN ON ICE). CRYSTALLIZATION (HANGING DROP VAPOR PHASE EQUILIBRATION) WAS ACHIEVED BY MIXING WITH AN EQUAL VOLUME OF THE PROTEIN COMPLEX WITH A RESERVOIR SOLUTION CONSISTING OF 70% 2-METHYL-2, 4-PENTANEDIOL (MPD) AND 100 MM HEPES (PH 6.5), AND SUSPENDING THE MIXTURE OVER THE RESERVOIR AT 277 K. SMALL ROD-LIKE CRYSTALS (0.01 X 0.01 X 0.05 MM) GREW WITHIN 2 DAYS. CRYSTALS WERE FLASH-COOLED DIRECTLY FROM THE DROP IN LIQUID N2. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.785 / Wavelength: 0.785 Å / Beamline: BL9-1 / Wavelength: 0.785 / Wavelength: 0.785 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 20, 2000 / Details: MIRRORS |
| Radiation | Monochromator: SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.785 Å / Relative weight: 1 |
| Reflection | Resolution: 1.89→15 Å / Num. obs: 15319 / % possible obs: 98.2 % / Observed criterion σ(I): -10 / Redundancy: 4.6 % / Biso Wilson estimate: 24.8 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 1.89→1.95 Å / Redundancy: 4.1 % / Rmerge(I) obs: 0.54 / Mean I/σ(I) obs: 2.7 / % possible all: 90.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RX1 Resolution: 1.89→14.83 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.94 / SU B: 6.352 / SU ML: 0.104 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.15 / ESU R Free: 0.141 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.52 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.89→14.83 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|