 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vbf: The holostructure of the branched-chain keto acid decarboxylase (... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vbf | ||||||
|---|---|---|---|---|---|---|---|
| Title | The holostructure of the branched-chain keto acid decarboxylase (KdcA) from Lactococcus lactis | ||||||
 Components Components | BRANCHED-CHAIN ALPHA-KETOACID DECARBOXYLASE | ||||||
 Keywords Keywords | LYASE / KDCA / FLAVOPROTEIN / THDP-DEPENDENT ENZYMES / THIAMINE PYROPHOSPHATE | ||||||
| Function / homology |  Function and homology information Function and homology informationbranched-chain-2-oxoacid decarboxylase activity / : / pyruvate decarboxylase activity / branched-chain amino acid biosynthetic process / thiamine pyrophosphate binding / magnesium ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  LACTOCOCCUS LACTIS (lactic acid bacteria) LACTOCOCCUS LACTIS (lactic acid bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Berthold, C.L. / Gocke, D. / Wood, M.D. / Leeper, F. / Pohl, M. / Schneider, G. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2007 Title: Crystal Structure of the Branched-Chain Keto Acid Decarboxylase (Kdca) from Lactococcus Lactis Provides Insights Into the Structural Basis for the Chemo- and Enantioselective Carboligation Reaction Authors: Berthold, C.L. / Gocke, D. / Wood, M.D. / Leeper, F. / Pohl, M. / Schneider, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vbf.cif.gz | 252.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vbf.ent.gz | 200 KB | Display | PDB format |
| PDBx/mmJSON format | 2vbf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vb/2vbfftp://data.pdbj.org/pub/pdb/validation_reports/vb/2vbf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2vbgC  1ovmS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper:
|
-Components
| #1: Protein | Mass: 63406.539 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: CHAIN A AND B CONTAIN THE COMPLETE ENZYME SEQUENCE AND SHORT N-TERMINAL TAGS WITH LINKERS, WHICH ARE BOUND IN ADJACENT DIMERS IN THE CRYSTAL. Source: (gene. exp.) LACTOCOCCUS LACTIS (lactic acid bacteria)Plasmid: PET28A / Production host: #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 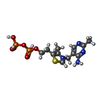  Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H19N4O7P2S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1109 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1109 / Source method: isolated from a natural source / Formula: H2OSequence details | THE PROTEIN CONTAINS A 23 AMINO ACID N-TERMINAL EXTENSION, MGSSHHHHHH | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.9 Å3/Da / Density % sol: 34.71 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 / Beamline: ID14-1 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Mar 3, 2007 |
| Radiation | Monochromator: DIAMOND (111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→56.17 Å / Num. obs: 127559 / % possible obs: 92.4 % / Observed criterion σ(I): 6 / Redundancy: 3.8 % / Biso Wilson estimate: 18 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 11.3 |
| Reflection shell | Resolution: 1.6→1.69 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.48 / Mean I/σ(I) obs: 2.2 / % possible all: 63.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1OVM Resolution: 1.6→30 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.951 / SU B: 1.674 / SU ML: 0.059 / Cross valid method: THROUGHOUT / ESU R: 0.094 / ESU R Free: 0.095 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE RESIDUES 182-187 HAVE NOT BEEN MODELED DUE TO LACK OF ELECTRON DENSITY THE FLEXIBLE REGION B 538-547 WAS MODELED AT OCCUPANCY 0.5
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.89 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|