 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2v42: Crystal structure of RseB: a sensor for periplasmic stress respon... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2v42 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of RseB: a sensor for periplasmic stress response in E. coli | ||||||
 Components Components | SIGMA-E FACTOR REGULATORY PROTEIN RSEB | ||||||
 Keywords Keywords | REGULATOR / REGULATORY PROTEIN / LIPOPROTEIN BINDING / SENSOR FOR PERIPLASMIC STRESS | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of polysaccharide biosynthetic process / antisigma factor binding / sigma factor antagonist complex / outer membrane-bounded periplasmic space / protein stabilization / negative regulation of DNA-templated transcription / lipid binding / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.75 Å | ||||||
 Authors Authors | Wollmann, P. / Zeth, K. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2007 Title: The Structure of Rseb: A Sensor in Periplasmic Stress Response of E. Coli. Authors: Wollmann, P. / Zeth, K. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2v42.cif.gz | 124.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2v42.ent.gz | 97.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2v42.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v4/2v42ftp://data.pdbj.org/pub/pdb/validation_reports/v4/2v42 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
NCS oper: (Code: given Matrix: (-0.45228, 0.89185, -0.00656), Vector: |
-Components
| #1: Protein | Mass: 34359.891 Da / Num. of mol.: 2 / Fragment: RESIDUES 23-318 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 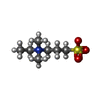  Mass: 195.280 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H17NO3S Mass: 195.280 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H17NO3S#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 44 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 44 / Source method: isolated from a natural source / Formula: H2OSequence details | RESIDUE 22 IS A METHIONINE WHICH WAS CLONED THERE TO HAVE A START CODON. RESIDUES 319-324 DO NOT ...RESIDUE 22 IS A METHIONINE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4 Å3/Da / Density % sol: 69.3 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7 Details: PROTEIN WAS CRYSTALLIZED FROM 2.4 M SODIUM MALONATE PH 7, 0.3 M DIMETHYLETHYLAMMONIUM PROPANE SULFONATE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 0.979 / Beamline: X10SA / Wavelength: 0.979 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jun 19, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 2.75→2.9 Å / Num. obs: 28941 / % possible obs: 97.5 % / Observed criterion σ(I): 2.4 / Redundancy: 12.4 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 18.8 |
| Reflection shell | Resolution: 2.75→2.9 Å / Redundancy: 12 % / Rmerge(I) obs: 0.51 / Mean I/σ(I) obs: 2.4 / % possible all: 92.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR Starting model: NONE Resolution: 2.75→20 Å / Cor.coef. Fo:Fc: 0.936 / Cor.coef. Fo:Fc free: 0.915 / SU B: 24.37 / SU ML: 0.232 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.418 / ESU R Free: 0.292 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 64.79 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|