- PDB-2v0r: crystal structure of a hairpin exchange variant (LTx) of the targ... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2v0r
Title
crystal structure of a hairpin exchange variant (LTx) of the targeting LINE-1 retrotransposon endonuclease
Components
LTX
Keywords
TRANSCRIPTION / APE-1 TYPE / ENDONUCLEASE / RETROTRANSPOSITION / RETROTRANSPOSON / PROTEIN ENGINEERING
Function / homology
Function and homology information
nucleic acid metabolic process / retrotransposition / type II site-specific deoxyribonuclease activity / Hydrolases; Acting on ester bonds; Endodeoxyribonucleases producing 5'-phosphomonoesters / RNA-directed DNA polymerase / RNA-directed DNA polymerase activity / DNA recombination / RNA binding / metal ion binding Similarity search - Function
Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O
Sequence details
THIS STRUCTURE IS A VARIANT OF THE PDB ENTRY 1VYB: L1 ENDONUCLEASE DOMAIN. THE LOOP REGION 193-201 ...THIS STRUCTURE IS A VARIANT OF THE PDB ENTRY 1VYB: L1 ENDONUCLEASE DOMAIN. THE LOOP REGION 193-201 IS REPLACED WITH A LOOP BELONGING TO RELATED ENDONUCLEASE.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.26 Å3/Da / Density % sol: 45.06 % / Description: NONE
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj

 Assembly
Assembly
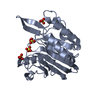
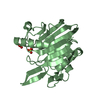


 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID23-1 / Wavelength: 1.072
/ Beamline: ID23-1 / Wavelength: 1.072  Processing
Processing