carbohydrate catabolic process / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme activity Similarity search - Function
Glycoside hydrolase, family 25 subgroup / Glycosyl hydrolases family 25 / Glycoside hydrolase, family 25 / Glycosyl hydrolases family 25 / Glycosyl hydrolase family 25 (GH25) domain profile. / Glycosidases / Glycoside hydrolase superfamily / TIM Barrel / Alpha-Beta Barrel / Alpha Beta Similarity search - Domain/homology
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 10-STRANDED BARREL THIS IS REPRESENTED BY A 11-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Mass: 18.015 Da / Num. of mol.: 372 / Source method: isolated from a natural source / Formula: H2O
-
Details
Sequence details
THE N-TERMINAL SIGNAL PEPTIDE, (SEQUENCE MKKKLFIRGIFILVSVMSVVAYLVFQGIFIPNQISA WAS REPLACED BY A ...THE N-TERMINAL SIGNAL PEPTIDE, (SEQUENCE MKKKLFIRGIFILVSVMSVVAYLVFQGIFIPNQISA WAS REPLACED BY A VECTOR DERIVED HEXAHISTIDINE TAG, (SEQUENCE MGSSHHHHHHM).
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.1 Å3/Da / Density % sol: 61 % / Description: NONE
Crystal grow
Method: vapor diffusion, hanging drop / pH: 8.5 / Details: 2.1M NH4 SO4, 0.1M TRIS PH 8.5.
Resolution: 1.4→33.73 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.947 / SU B: 0.832 / SU ML: 0.034 / Cross valid method: THROUGHOUT / ESU R: 0.059 / ESU R Free: 0.061 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.21217
3191
5.1 %
RANDOM
Rwork
0.18733
-
-
-
obs
0.18859
59675
99.76 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly

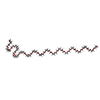




 Mass: 1529.829 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C69H140O35 / Comment: precipitant*YM
Mass: 1529.829 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C69H140O35 / Comment: precipitant*YM Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing