Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.61 Å3/Da / Density % sol: 65.94 %
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.8 Details: DROPS WERE FORMED BY MIXING EQUAL AMOUNTS OF 18-24 MG/ML PROTEIN IN 100 MM HEPES PH 7.0, 400 MM NACL, 30% GLYCEROL, 1 MM EDTA AND 10 MM N,N- DIMETHYLUNDECYLAMIN-N-OXIDE (C11DAO) WITH A ...Details: DROPS WERE FORMED BY MIXING EQUAL AMOUNTS OF 18-24 MG/ML PROTEIN IN 100 MM HEPES PH 7.0, 400 MM NACL, 30% GLYCEROL, 1 MM EDTA AND 10 MM N,N- DIMETHYLUNDECYLAMIN-N-OXIDE (C11DAO) WITH A PRECIPITANT SOLUTION OF 0.1 M ACETATE PH 4.8 40 MM C11DAO, 20.8 MM N,-DIMETHYLDECYLAMINE-N-OXIDE (DDAO), 2 MM DIHYDROOROTATE (DHO) THE HANGING DROPS WERE INCUBATED AGAINST 0.5 ML RESERVOIR OF 0.1 M ACETATE PH 4.8, 1.6-2.2 M AMMONIUM SULFATE AND 30% GLYCEROL., VAPOR DIFFUSION, HANGING DROP, temperature 293K
-
Data collection
Diffraction
ID
Mean temperature (K)
Crystal-ID
1
100
1
2
1
Diffraction source
Source: SYNCHROTRON / Site: MAX II / Beamline: I711 / Wavelength: 1.092 Å
Detector
Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Apr 27, 2004 / Details: mirrors
Radiation
ID
Protocol
Monochromatic (M) / Laue (L)
Scattering type
Wavelength-ID
1
SINGLEWAVELENGTH
M
x-ray
1
2
SINGLEWAVELENGTH
M
x-ray
1
Radiation wavelength
Wavelength: 1.092 Å / Relative weight: 1
Reflection
Resolution: 2.1→18.83 Å / Num. obs: 33570 / % possible obs: 98 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 22.487 Å2 / Rmerge(I) obs: 0.114 / Net I/σ(I): 12.17
Reflection shell
Diffraction-ID: 1,2
Resolution (Å)
Highest resolution (Å)
Rmerge(I) obs
Mean I/σ(I) obs
Num. measured obs
Num. unique all
% possible all
2.1-2.2
0.327
5.8
26848
4367
99.5
2.2-2.3
0.271
6.7
22493
3643
99.2
2.3-2.4
0.255
7.1
18935
3055
99.1
2.4-2.6
0.208
8.2
29772
4785
98.9
2.6-2.7
0.173
9.6
11800
1900
98.9
2.7-3
0.14
10.9
26614
4269
98.4
3-4
0.082
16.6
41760
6665
97.6
4-5
0.058
24
14896
2381
96.4
5-6
0.061
22.8
6695
1065
95.6
6
0.047
28.6
8752
1440
93.4
-
Processing
Software
Name
Version
Classification
NB
REFMAC
5.2.0019
refinement
REFMAC
5.2.0019
refinement
REFMAC
5.2.0019
refinement
MAR345dtb
datacollection
XDS
datareduction
XSCALE
datascaling
CNS
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1d3g
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj
 Assembly
Assembly





 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2
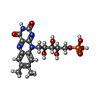
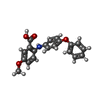
Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P
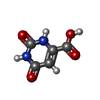
Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 156.096 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H4N2O4
Mass: 156.096 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H4N2O4 Mass: 335.353 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H17NO4
Mass: 335.353 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H17NO4 Mass: 201.349 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H27NO
Mass: 201.349 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H27NO Sample preparation
Sample preparation / Beamline: I711 / Wavelength: 1.092 Å
/ Beamline: I711 / Wavelength: 1.092 Å Processing
Processing