 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
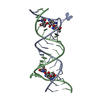
| Entry | Database: PDB / ID: 2pn3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Hepatitis C Virus IRES Subdomain IIa | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA / HCV / IRES / Loop IIa / Magnesium / Crystallization | ||||||
| Function / homology | RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.9 Å | ||||||
 Authors Authors | Zhao, Q. / Han, Q. / Kissinger, C.R. / Hermann, T. / Thompson, P.A. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2008 Title: Structure of hepatitis C virus IRES subdomain IIa. Authors: Zhao, Q. / Han, Q. / Kissinger, C.R. / Hermann, T. / Thompson, P.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2pn3.cif.gz | 33.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2pn3.ent.gz | 23.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2pn3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pn/2pn3ftp://data.pdbj.org/pub/pdb/validation_reports/pn/2pn3 | HTTPS FTP |
|---|
-Related structure data








-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 7644.589 Da / Num. of mol.: 1 / Fragment: HCV IRES Subdomain IIa (residue 47-70) / Mutation: G47C, U48G, A70C / Source method: obtained synthetically Details: This sequence occurs naturally in hepatitis C virus |
|---|---|
| #2: RNA chain | Mass: 6564.623 Da / Num. of mol.: 1 / Fragment: HCV IRES Subdomain IIa (residue 98-117) / Mutation: A99C, A116G / Source method: obtained synthetically Details: This sequence occurs naturally in hepatitis C virus |
| #3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 51.74 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: evaporation / pH: 6.2 Details: 5% MPD, 5mM MgSO4, 12.5mM NaCl, 50mM KCl, 25mM Cacodylate equilibrated with 65%MPD, pH 6.2, EVAPORATION, temperature 290.0K | ||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 77 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-ID-B / Wavelength: 0.9203, 0.9197, 0.90686 / Beamline: 14-ID-B / Wavelength: 0.9203, 0.9197, 0.90686 | ||||||||||||
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Oct 30, 2004 / Details: mirrors | ||||||||||||
| Radiation | Monochromator: double-crystal Si 111 / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.9→50 Å / Num. all: 3650 / Num. obs: 3420 / % possible obs: 93.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 11.2 % / Biso Wilson estimate: 55.3 Å2 / Rmerge(I) obs: 0.081 / Net I/σ(I): 27.5 | ||||||||||||
| Reflection shell | Resolution: 2.9→3 Å / Redundancy: 8.1 % / Rmerge(I) obs: 0.345 / Mean I/σ(I) obs: 4.6 / Num. unique all: 253 / % possible all: 75.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.9→45 Å / Cor.coef. Fo:Fc: 0.891 / Cor.coef. Fo:Fc free: 0.843 / SU B: 18.57 / SU ML: 0.349 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.46 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.297 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→45 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→2.977 Å / Total num. of bins used: 20
|
