 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2p6n | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human DEAD-box RNA helicase DDX41, helicase domain | ||||||
 Components Components | ATP-dependent RNA helicase DDX41 | ||||||
 Keywords Keywords | HYDROLASE / RNA / HELICASE / DEAD / STRUCTURAL GENOMICS / STRUCTURAL GENOMICS CONSORTIUM / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationSTING mediated induction of host immune responses / IRF3-mediated induction of type I IFN / catalytic step 2 spliceosome / Regulation of innate immune responses to cytosolic DNA / mRNA Splicing - Major Pathway / spliceosomal complex / mRNA splicing, via spliceosome / cell population proliferation / RNA helicase activity / cell differentiation ...STING mediated induction of host immune responses / IRF3-mediated induction of type I IFN / catalytic step 2 spliceosome / Regulation of innate immune responses to cytosolic DNA / mRNA Splicing - Major Pathway / spliceosomal complex / mRNA splicing, via spliceosome / cell population proliferation / RNA helicase activity / cell differentiation / RNA helicase / mRNA binding / apoptotic process / ATP hydrolysis activity / RNA binding / nucleoplasm / ATP binding / membrane / nucleus / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Karlberg, T. / Ogg, D. / Arrowsmith, C.H. / Berglund, H. / Busam, R.D. / Collins, R. / Dahlgren, L.G. / Edwards, A. / Flodin, S. / Flores, A. ...Karlberg, T. / Ogg, D. / Arrowsmith, C.H. / Berglund, H. / Busam, R.D. / Collins, R. / Dahlgren, L.G. / Edwards, A. / Flodin, S. / Flores, A. / Graslund, S. / Hallberg, B.M. / Hammarstrom, M. / Johansson, I. / Kotenyova, T. / Lehtio, L. / Moche, M. / Nordlund, P. / Nyman, T. / Persson, C. / Sagemark, J. / Stenmark, P. / Sundstrom, M. / Thorsell, A.G. / Van Den Berg, S. / Weigelt, J. / Holmberg-Schiavone, L. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Plos One / Year: 2010 Title: Comparative Structural Analysis of Human DEAD-Box RNA Helicases. Authors: Schutz, P. / Karlberg, T. / van den Berg, S. / Collins, R. / Lehtio, L. / Hogbom, M. / Holmberg-Schiavone, L. / Tempel, W. / Park, H.W. / Hammarstrom, M. / Moche, M. / Thorsell, A.G. / Schuler, H. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1, 2 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 2 ... BIOMOLECULE: 1, 2 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 2 CHAIN(S). AUTHORS STATE THAT THE BIOLOGICAL UNIT OF THIS PROTEIN IS A MONOMER. SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2p6n.cif.gz | 74.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2p6n.ent.gz | 56 KB | Display | PDB format |
| PDBx/mmJSON format | 2p6n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2p6n_validation.pdf.gz | 442.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2p6n_full_validation.pdf.gz | 449.4 KB | Display | |
| Data in XML | 2p6n_validation.xml.gz | 13.7 KB | Display | |
| Data in CIF | 2p6n_validation.cif.gz | 17.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p6/2p6nftp://data.pdbj.org/pub/pdb/validation_reports/p6/2p6n | HTTPS FTP |
-Related structure data
| Related structure data | 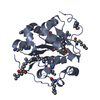 2g9nC  2pl3C  2rb4C  3b7gC  3berC  3borC  3dkpC  3fe2C  3iuyC  3ly5C  2i4iS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: LEU / Beg label comp-ID: LEU / End auth comp-ID: VAL / End label comp-ID: VAL / Refine code: 5 / Auth seq-ID: 406 - 565 / Label seq-ID: 28 - 187
| ||||||||||||||||||
| Details | The biological unit is a monomer, the asymmetric unit contains two monomers |
-Components
| #1: Protein | Mass: 21172.273 Da / Num. of mol.: 2 / Fragment: HELICASE DOMAIN / Mutation: R525C Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DDX41, ABS / Plasmid: pNIC-Bsa4 / Production host:  References: UniProt: Q9UJV9, Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 48.93 % |
|---|---|
| Crystal grow | Temperature: 293 K / pH: 5.5 Details: 25% PEG 3350, 200mM Lithium sulfate, 100mM Bis-Tris, pH 5.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K, pH 5.50 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 1.04005 / Beamline: ID14-4 / Wavelength: 1.04005 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 29, 2006 Details: DOUBLE CRYSTAL, SI(111) OR SI(311), TOROIDAL MIRROR |
| Radiation | Monochromator: KHOZU, WITH A MCLENNON CONTROLLER CONTAINING A LN2 COOLED SI111 CRYSTAL Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.04005 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→30 Å / Num. obs: 13827 / % possible obs: 99.9 % / Observed criterion σ(I): 0 / Redundancy: 40 % / Rmerge(I) obs: 0.103 / Rsym value: 0.029 / Net I/σ(I): 35.9 |
| Reflection shell | Resolution: 2.6→2.8 Å / Redundancy: 41.8 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 12.6 / Rsym value: 0.084 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2I4I Resolution: 2.6→29.72 Å / Cor.coef. Fo:Fc: 0.932 / Cor.coef. Fo:Fc free: 0.907 / SU B: 27.859 / SU ML: 0.266 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.559 / ESU R Free: 0.337 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 47.341 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→29.72 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.667 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
