 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ozr: MMP13 Catalytic Domain Complexed with 4-{[1-methyl-2,4-dioxo-6-(3... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ozr | ||||||
|---|---|---|---|---|---|---|---|
| Title | MMP13 Catalytic Domain Complexed with 4-{[1-methyl-2,4-dioxo-6-(3-phenylprop-1-yn-1-yl)-1,4-dihydroquinazolin-3(2H)-yl]methyl}benzoic acid | ||||||
 Components Components | Collagenase 3 | ||||||
 Keywords Keywords | HYDROLASE / Crystal Complex Structure / Matrix Metalloproteinase / MMP13 Catalytic Domain / MMP13 Specific Inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationendochondral ossification / RUNX2 regulates genes involved in cell migration / Hydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases / bone morphogenesis / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / bone mineralization / response to amyloid-beta / Collagen degradation / collagen catabolic process ...endochondral ossification / RUNX2 regulates genes involved in cell migration / Hydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases / bone morphogenesis / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / bone mineralization / response to amyloid-beta / Collagen degradation / collagen catabolic process / extracellular matrix disassembly / Degradation of the extracellular matrix / collagen binding / extracellular matrix organization / metalloendopeptidase activity / extracellular matrix / endopeptidase activity / serine-type endopeptidase activity / calcium ion binding / proteolysis / : / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Johnson, A.R. / Pavlovsky, A.G. / Ortwine, D.F. / Prior, F. / Man, C.-F. / Bornemeier, D.A. / Banotai, C.A. / Mueller, W.T. / McConnell, P. / Yan, C.H. ...Johnson, A.R. / Pavlovsky, A.G. / Ortwine, D.F. / Prior, F. / Man, C.-F. / Bornemeier, D.A. / Banotai, C.A. / Mueller, W.T. / McConnell, P. / Yan, C.H. / Baragi, V. / Lesch, C. / Roark, W.H. / Lie, J.J. / Fasquelle, V. / Wilson, M. / Robertson, D. / Datta, K. / Guzman, R. / Han, H.-K. / Dyer, R.D. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. Authors: Johnson, A.R. / Pavlovsky, A.G. / Ortwine, D.F. / Prior, F. / Man, C.F. / Bornemeier, D.A. / Banotai, C.A. / Mueller, W.T. / McConnell, P. / Yan, C. / Baragi, V. / Lesch, C. / Roark, W.H. / ...Authors: Johnson, A.R. / Pavlovsky, A.G. / Ortwine, D.F. / Prior, F. / Man, C.F. / Bornemeier, D.A. / Banotai, C.A. / Mueller, W.T. / McConnell, P. / Yan, C. / Baragi, V. / Lesch, C. / Roark, W.H. / Wilson, M. / Datta, K. / Guzman, R. / Han, H.K. / Dyer, R.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ozr.cif.gz | 305 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ozr.ent.gz | 247 KB | Display | PDB format |
| PDBx/mmJSON format | 2ozr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oz/2ozrftp://data.pdbj.org/pub/pdb/validation_reports/oz/2ozr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ow9SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Refine code: 4
|
-Components
-Protein , 1 types, 8 molecules ABCDEFGH
| #1: Protein | Mass: 19069.314 Da / Num. of mol.: 8 / Fragment: Catalytic Domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MMP13 / Plasmid: pGEMEX1 / Production host:  References: UniProt: P45452, Hydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases |
|---|
-Non-polymers , 5 types, 1076 molecules 

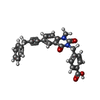
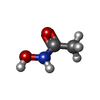

| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: Zn / Details: Special chemical synthesis Mass: 65.409 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: Zn / Details: Special chemical synthesis#3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Ca / Details: commersially synthesised Mass: 40.078 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Ca / Details: commersially synthesised#4: Chemical | ChemComp-GG1 /  Mass: 424.448 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C26H20N2O4 Mass: 424.448 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C26H20N2O4#5: Chemical | ChemComp-HAE /  Mass: 75.067 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H5NO2 / Comment: medication*YM Mass: 75.067 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H5NO2 / Comment: medication*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1024 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.44 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: Dissolved in DMSO inhibitor was mixed with protein (protein concentration: 1mg/ml) at 5:1 ratio, 0.1 M acetohydrohamic acid added. Ternary complex was concentrated (to 17 mg/ml protien ...Details: Dissolved in DMSO inhibitor was mixed with protein (protein concentration: 1mg/ml) at 5:1 ratio, 0.1 M acetohydrohamic acid added. Ternary complex was concentrated (to 17 mg/ml protien concentration). 1-2 uL hagind drops were mixed with same amount of reservoit solution (2.1 M ammonium sulfate in 0.1 M Hepes buffer) , pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Nov 25, 2001 / Details: mirrors |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→50 Å / Num. all: 160245 / Num. obs: 69624 / % possible obs: 90.7 % / Redundancy: 2.3 % / Biso Wilson estimate: 26 Å2 / Rsym value: 0.096 / Χ2: 1.433 / Net I/σ(I): 6.5 |
| Reflection shell | Resolution: 2.1→2.18 Å / Mean I/σ(I) obs: 2 / Num. unique all: 4793 / Rsym value: 0.342 / Χ2: 0.991 / % possible all: 62.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB Entry 2OW9 Resolution: 2.3→50 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.898 / SU B: 18.324 / SU ML: 0.264 / TLS residual ADP flag: LIKELY RESIDUAL / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.841 / ESU R Free: 0.38 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: Used NCS Restraints
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.737 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→50 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 1279 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 20.4251 Å / Origin y: 0.0314 Å / Origin z: 28.1447 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Refine-ID: X-RAY DIFFRACTION / Refine TLS-ID: 1 / Selection: ALL
|
