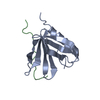
Chemotaxis Inhibitory Protein of Staphylococcus Aureus (CHIPS) complexed to the N-terminus (7-28) ...Chemotaxis Inhibitory Protein of Staphylococcus Aureus (CHIPS) complexed to the N-terminus (7-28) of the C5aR receptor.
Mass: 10475.967 Da / Num. of mol.: 1 / Fragment: Chemotaxis inhibiting protein CHIPS(59-149). Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus subsp. aureus str. Newman (bacteria) Gene: chp / Plasmid: pRSET B / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 / Variant (production host): DE3 / References: UniProt: A6QIG7
#2: Protein/peptide
C5aanaphylatoxinchemotacticreceptor1
Mass: 2710.813 Da / Num. of mol.: 1 / Fragment: C5aR(P7-28S) / Source method: obtained synthetically Details: Fmoc/tBu-based peptide synthesis. Sulfated tyrosines 11 and 14 were introduced as Fmoc-2-chlorotrityl protected building blocks. Source: (synth.) Homo sapiens (human) / References: UniProt: P21730
Compound details
AMBIGUOUS CONFORMATIONAL STATES IN SLOW EXCHANGE ARE PRESENT FOR RESIDUES PRO25-VAL26-ASP27-LYS28 ...AMBIGUOUS CONFORMATIONAL STATES IN SLOW EXCHANGE ARE PRESENT FOR RESIDUES PRO25-VAL26-ASP27-LYS28 OF THE C5AR(P7-28S) PEPTIDE WHEN PRESENT IN THE COMPLEX, PROBABLY CAUSED BY CIS-TRANS ISOMERIZATION AT PRO25. THE RATIO OF THE TWO SLOWLY EXCHANGING CONFORMERS IS ABOUT 1:1. CHEMICAL SHIFTS OF RESIDUES IN CONFORMER A ARE ATTRIBUTED TO RESIDUES 25-28. CHEMICAL SHIFTS OF CONFORMER B ARE DESCRIBED BY RESIDUES 125-128 (OR 25'-28'). RESIDUES 25-28 OF THE C5AR(P7-28S) PEPTIDE ARE NOT IN DIRECT CONTACT TO THE CHIPS PROTEIN, ACCORDING TO EXPERIMENTAL NMR DATA, AND DO NOT CONTRIBUTE TO SPECIFIC BINDING PROPERTIES OF THE CHIPS COMPLEX. PEPTIDE N-TERMINAL END BLOCKED BY AN ACETYL GROUP. PEPTIDE C-TERMINAL END BLOCKED BY A NH2 GROUP. CONTAINS TWO SULFATED TYROSINES (TYS) AT POSITIONS 11 AND 14.
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: SOLUTION NMR Details: Chemotaxis Inhibitory Protein of Staphylococcus Aureus (CHIPS) complexed to the N-terminus (7-28) of the C5aR receptor.
NMR experiment
Conditions-ID
Experiment-ID
Solution-ID
Type
1
1
1
2D 1H-15N HSQC
1
2
1
2D 1H-15N HSQC
1
3
1
2D 1H-15N HSQC
2
4
2
2D 1H-13C HSQC
2
5
2
2D 1H-13C HSQC
2
6
2
2D 1H-13C HSQC
2
7
2
3DCBCA(CO)NH
2
8
2
3D HN(CA)CB
2
9
2
3D HNCO
2
10
2
3DHBHACBCA(CO)NH
2
11
2
3D (H)CCH-TOCSY
2
12
2
3D (H)CCH-TOCSY
1
13
1
3D 1H-15N NOESY
2
14
2
3D 1H-15N NOESY
1
15
1
3D 1H-15N TOCSY
2
16
2
3D 1H-13C NOESY
2
17
2
3D 1H-13C NOESY
2
18
2
2D 1H-1H NOESY
2
19
2
2D 1H-1H TOCSY
2
20
2
3DCOCA(HN)
2
21
2
3D-CNH-NOESY
2
22
2
2D-HBHD aromatic
2
23
2
2D-HBHE aromatic
2
24
2
2D-CBHD aromatic
2
25
2
2D-CBHE aromatic
2
26
2
3D HNHA
2
27
2
3D HNHB
NMR details
Text: STANDARD TRIPLE RESONANCE EXPERIMENTS WERE USED FOR ASSIGNMENT AND STRUCTURE DETERMINATION OF THE CHIPS PROTEIN. THE UNLABELLED PEPTIDE C5AR(P7-28S) IN THE COMPLEX HAS BEEN SOLVED BY MEANS OF ...Text: STANDARD TRIPLE RESONANCE EXPERIMENTS WERE USED FOR ASSIGNMENT AND STRUCTURE DETERMINATION OF THE CHIPS PROTEIN. THE UNLABELLED PEPTIDE C5AR(P7-28S) IN THE COMPLEX HAS BEEN SOLVED BY MEANS OF ISOTOPE-FILTERED 2D SPECTRA. TO EXTRACT INTERMOLECULAR NOE'S BETWEEN [15N,13C] LABELLED CHIPS PROTEIN AND UNLABELLED C5AR(P7-28S) PEPTIDE, SEVERAL 2D-13C-FILTERED NOESY AND 3D 13C-EDITED-13C-FILTERED NOESY SPECTRA WERE RECORDED AT 900 MHZ. FOR THE 3D FILTERED SPECTRA THE 13C-HSQC DETECTION STEP WAS OPTIMIZED BY RECORDING TWO SPECTRA, ONE WITH THE 13C CARRIER FREQUENCY PLACED IN THE AROMATIC REGION, AND ONE WITH THE 13C CARRIER FREQUENCY SET TO THE ALIFATIC REGION. THE MIXING TIME USED WAS 200 MS, TO FORCE A GOOD SENSITIVITY NECESSARY FOR THE COLLECTION OF A SUFFICIENT NUMBER OF INTERMOLECULAR NOE'S.
-
Sample preparation
Details
Solution-ID
Contents
Solvent system
1
0.5 mM [U-99% 15N] protein, 0.5 mM entity_2, 20 mM sodium phosphate, 0.1 % sodium azide, 90 % H2O, 10 % D2O, 90% H2O/10% D2O
90% H2O/10% D2O
2
1.0 mM [U-99% 13C; U-99% 15N] protein, 1.0 mM entity_2, 20 mM sodium phosphate, 0.1 % sodium azide, 90 % H2O, 10 % D2O, 90% H2O/10% D2O
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Staphylococcus aureus subsp. aureus str. Newman (bacteria)
Staphylococcus aureus subsp. aureus str. Newman (bacteria) Homo sapiens (human)
Homo sapiens (human) Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj











 Assembly
Assembly
 HSQC
HSQC Sample preparation
Sample preparation Processing
Processing