 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2jg0: Family 37 trehalase from Escherichia coli in complex with 1- thia... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2jg0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Family 37 trehalase from Escherichia coli in complex with 1- thiatrehazolin | |||||||||
 Components Components | PERIPLASMIC TREHALASE | |||||||||
 Keywords Keywords | HYDROLASE / FAMILY 37 / INHIBITOR / TREHALASE / GLYCOSIDE HYDROLASE / PERIPLASMIC / GLYCOSIDASE / 1-THIATREHAZOLIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationalpha,alpha-trehalase / alpha,alpha-trehalase activity / trehalose catabolic process / cellular hyperosmotic response / outer membrane-bounded periplasmic space / DNA damage response Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | |||||||||
 Authors Authors | Gibson, R.P. / Gloster, T.M. / Roberts, S. / Warren, R.A.J. / Storch De Gracia, I. / Garcia, A. / Chiara, J.L. / Davies, G.J. | |||||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2007 Title: Molecular Basis for Trehalase Inhibition Revealed by the Structure of Trehalase in Complex with Potent Inhibitors. Authors: Gibson, R.P. / Gloster, T.M. / Roberts, S. / Warren, R.A.J. / Storch De Gracia, I. / Garcia, A. / Chiara, J.L. / Davies, G.J. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2jg0.cif.gz | 246.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2jg0.ent.gz | 195.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2jg0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jg/2jg0ftp://data.pdbj.org/pub/pdb/validation_reports/jg/2jg0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2jf4SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60526.531 Da / Num. of mol.: 1 / Fragment: RESIDUES 31-565 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Sugar | ChemComp-TTZ / 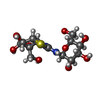  Type: D-saccharide / Mass: 382.387 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H22N2O9S Type: D-saccharide / Mass: 382.387 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H22N2O9S |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 676 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 676 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THE FIRST 30 RESIDUES CORRESPOND TO A SIGNAL PEPTIDE THAT IS PREDICTED TO HAVE CLEAVED DURING ...THE FIRST 30 RESIDUES CORRESPOND |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 43 % |
|---|---|
| Crystal grow | pH: 6.5 Details: 15 MG/ML PROTEIN, 25% PEG3350 AND 0.1 M BIS-TRIS HCL, PH 6.5. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.97377 / Beamline: ID29 / Wavelength: 0.97377 |
| Detector | Type: ADSC CCD / Detector: CCD / Details: TOROIDAL MIRROR |
| Radiation | Monochromator: SI(311) AND SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97377 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→30 Å / Num. obs: 83167 / % possible obs: 100 % / Redundancy: 5.3 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 12.6 |
| Reflection shell | Resolution: 1.5→1.58 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 4 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PBD ENTRY 2JF4 Resolution: 1.5→28.36 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.942 / SU B: 2.372 / SU ML: 0.042 / Cross valid method: THROUGHOUT / ESU R: 0.086 / ESU R Free: 0.072 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. NO ELECTRON DENSITY CAN BE OBSERVED FOR THE CHAIN BETWEEN 102 AND 107. THERE IS DISORDERED DENSITY IN THE REGION OF 478,546,547 WHICH HAS ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. NO ELECTRON DENSITY CAN BE OBSERVED FOR THE CHAIN BETWEEN 102 AND 107. THERE IS DISORDERED DENSITY IN THE REGION OF 478,546,547 WHICH HAS BEEN MODELLED AS BEST AS POSSIBLE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 8.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→28.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|