HELIX THE FOLLOWING HELIX RECORDS HAVE BEEN REMOVED AT AUTHORS REQUEST: HELIX 5 5 ILE A 82 GLN A ... HELIX THE FOLLOWING HELIX RECORDS HAVE BEEN REMOVED AT AUTHORS REQUEST: HELIX 5 5 ILE A 82 GLN A 84 5 HELIX 9 9 GLY A 197 ALA A 199 5
Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O
-
Details
Compound details
ENGINEERED RESIDUE IN CHAIN A, ASN 109 TO GLN ENGINEERED RESIDUE IN CHAIN A, ASN 185 TO GLN
Has protein modification
Y
Sequence details
THE PROTEIN SEQUENCE ALIGNS WITH UNIPROT ENTRY O09008. THE SEQUENCE 42-44(GLY-PRO-MET) IN 2J0B IS ...THE PROTEIN SEQUENCE ALIGNS WITH UNIPROT ENTRY O09008. THE SEQUENCE 42-44(GLY-PRO-MET) IN 2J0B IS NOT PRESENT IN THE UNIPROT ENTRY BUT IS DERIVED FROM THE EXPRESSION VECTOR. IN ADDITION, THERE IS A MUTATION AT RESIDUE 185 (ASN TO GLN)
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.08 Å3/Da / Density % sol: 40.8 % Description: LIGAND SOAK, STRUCTURE OBTAINED THROUGH FOURIER DIFFERENCE
Crystal grow
Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: 0.2 M POTASSIUM SULFATE, 20% PEG 3350, pH 6.50
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly

 SPODOPTERA FRUGIPERDA (fall armyworm)
SPODOPTERA FRUGIPERDA (fall armyworm)


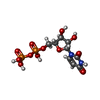

 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM
Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Sample preparation
Sample preparation / Beamline: X06SA / Wavelength: 0.9195
/ Beamline: X06SA / Wavelength: 0.9195  Processing
Processing