Journal: Mol Cell / Year: 2006 Title: Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Authors: Lisa Craig / Niels Volkmann / Andrew S Arvai / Michael E Pique / Mark Yeager / Edward H Egelman / John A Tainer / Abstract: Type IV pili (T4P) are long, thin, flexible filaments on bacteria that undergo assembly-disassembly from inner membrane pilin subunits and exhibit astonishing multifunctionality. Neisseria ...Type IV pili (T4P) are long, thin, flexible filaments on bacteria that undergo assembly-disassembly from inner membrane pilin subunits and exhibit astonishing multifunctionality. Neisseria gonorrhoeae (gonococcal or GC) T4P are prototypic virulence factors and immune targets for increasingly antibiotic-resistant human pathogens, yet detailed structures are unavailable for any T4P. Here, we determined a detailed experimental GC-T4P structure by quantitative fitting of a 2.3 A full-length pilin crystal structure into a 12.5 A resolution native GC-T4P reconstruction solved by cryo-electron microscopy (cryo-EM) and iterative helical real space reconstruction. Spiraling three-helix bundles form the filament core, anchor the globular heads, and provide strength and flexibility. Protruding hypervariable loops and posttranslational modifications in the globular head shield conserved functional residues in pronounced grooves, creating a surprisingly corrugated pilus surface. These results clarify T4P multifunctionality and assembly-disassembly while suggesting unified assembly mechanisms for T4P, archaeal flagella, and type II secretion system filaments.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Neisseria gonorrhoeae (bacteria)
Neisseria gonorrhoeae (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj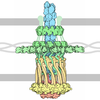

 Assembly
Assembly

 Mass: 148.200 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16O3
Mass: 148.200 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16O3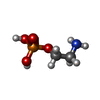
 Mass: 141.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H8NO4P
Mass: 141.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H8NO4P Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL7-1 / Wavelength: 1.08 Å
/ Beamline: BL7-1 / Wavelength: 1.08 Å Processing
Processing