 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2h5d: 0.9A resolution crystal structure of alpha-lytic protease complex... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2h5d | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 0.9A resolution crystal structure of alpha-lytic protease complexed with a transition state analogue, MeOSuc-Ala-Ala-Pro-Val boronic acid | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / A-LYTIC PROTEASE / SERINE PROTEASE / ACYLATION TRANSITION STATE / CATALYSIS / PROTEIN FOLDING / PROTEIN STABILITY / PACKING DISTORTION / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationalpha-lytic endopeptidase / serine-type endopeptidase activity / proteolysis / extracellular region Similarity search - Function | |||||||||
| Biological species |  Lysobacter enzymogenes (bacteria) Lysobacter enzymogenes (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / REFINEMENT OF PREVIOUSLY-SOLVED STRUCTURE OF ALPHA-LYTIC PROTEASE BOUND TO MEOSUC-ALA-ALA-PRO-ALA BORONIC ACID / Resolution: 0.9 Å X-RAY DIFFRACTION / SYNCHROTRON / REFINEMENT OF PREVIOUSLY-SOLVED STRUCTURE OF ALPHA-LYTIC PROTEASE BOUND TO MEOSUC-ALA-ALA-PRO-ALA BORONIC ACID / Resolution: 0.9 Å | |||||||||
 Authors Authors | Fuhrmann, C.N. / Agard, D.A. | |||||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2006 Title: Subangstrom crystallography reveals that short ionic hydrogen bonds, and not a His-Asp low-barrier hydrogen bond, stabilize the transition state in serine protease catalysis Authors: Fuhrmann, C.N. / Daugherty, M.D. / Agard, D.A. #1: Journal: J.Mol.Biol. / Year: 2004Title: The 0.83A resolution crystal structure of alpha-lytic protease reveals the detailed structure of the active site and identifies a source of conformational strain Authors: Fuhrmann, C.N. / Kelch, B.A. / Ota, N. / Agard, D.A. #2: Journal: Biochemistry / Year: 1989Title: Structural analysis of specificity: alpha-lytic protease complexes with analogues of reaction intermediates Authors: Bone, R. / Frank, D. / Kettner, C.A. / Agard, D.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2h5d.cif.gz | 153.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2h5d.ent.gz | 122.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2h5d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h5/2h5dftp://data.pdbj.org/pub/pdb/validation_reports/h5/2h5d | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2h5cC  1p02S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 19875.131 Da / Num. of mol.: 1 / Fragment: MATURE PROTEASE DOMAIN (RESIDUES 200-397) / Source method: isolated from a natural source / Details: gene alpha-LP / Source: (natural) Lysobacter enzymogenes (bacteria) / Secretion: SECRETED BY THE NATIVE BACTERIUM / References: UniProt: P00778, alpha-lytic endopeptidase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | 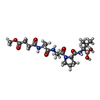  Type: Peptide-like / Class: Inhibitor / Mass: 470.325 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: METHOXYSUCCINYL-ALA-ALA-PRO-VALINE BORONIC ACID Type: Peptide-like / Class: Inhibitor / Mass: 470.325 Da / Num. of mol.: 1 / Source method: obtained synthetically / References: METHOXYSUCCINYL-ALA-ALA-PRO-VALINE BORONIC ACID | ||||||||
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 432 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 432 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | GLYCEROL BOUND TO THE CATALYTIC ADDUCT, CREATING A MIMIC OF THE ACYLATION TRANSITION | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.6 % |
|---|---|
| Crystal grow | Temperature: 298 K / pH: 8 Details: 1.3M LITHIUM SULFATE, 0.02M TRIS, PH 8.0, VAPOR DIFFUSION, HANGING DROP, TEMPERATURE 298K, pH 8.00 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.2 / Wavelength: 0.75 / Beamline: 8.2.2 / Wavelength: 0.75 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jun 19, 2004 |
| Radiation | Monochromator: DOUBLE CRYSTAL, SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.75 Å / Relative weight: 1 |
| Reflection | Resolution: 0.9→28.5 Å / Num. obs: 147100 / % possible obs: 99.9 % / Redundancy: 8.7 % / Rmerge(I) obs: 0.085 / Net I/σ(I): 27.9 |
| Reflection shell | Resolution: 0.9→0.91 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.448 / Mean I/σ(I) obs: 4.8 / % possible all: 99.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: REFINEMENT OF PREVIOUSLY-SOLVED STRUCTURE OF ALPHA-LYTIC PROTEASE BOUND TO MEOSUC-ALA-ALA-PRO-ALA BORONIC ACID Starting model: SAME PROTEIN BOUND TO MEOSUC-ALA-ALA-PRO-ALA BORONIC ACID AT 0.9A RESOLUTION (UNPUBLISHED DATA, BUT RELATED TO 1P02) Resolution: 0.9→20 Å / Num. parameters: 20128 / Num. restraintsaints: 23688 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH & HUBER Details: HYDROGEN ATOMS WERE INCLUDED IN THE REFINEMENT AS "RIDING HYDROGENS", WITH POSITION AND GEOMETRY FIXED TO THOSE VALUES DEFINED BY SHELXL-97. METHYL AND HYDROXYL HYDROGENS ON SINGLE-CONFORMER ...Details: HYDROGEN ATOMS WERE INCLUDED IN THE REFINEMENT AS "RIDING HYDROGENS", WITH POSITION AND GEOMETRY FIXED TO THOSE VALUES DEFINED BY SHELXL-97. METHYL AND HYDROXYL HYDROGENS ON SINGLE-CONFORMER SIDECHAINS WERE EACH POSITIONED AT A TORSION ANGLE THAT BEST SATISFIED POSITIVE DIFFERENCE ELECTRON DENSITY (USING INSTRUCTIONS HFIX 137 AND HFIX 147, RESPECTIVELY). IT SHOULD BE NOTED THAT THE LENGTH OF DONOR-HYDROGEN BONDS IN THIS STRUCTURE ARE LIKELY SHORTER THAN THEIR TRUE INTERNUCLEAR DISTANCE; THESE BOND LENGTHS ARE DEFINED BY SHELXL-97 PARAMETERS. THE POSITIONS OF SEVEN HYDROGEN ATOMS WERE ALLOWED TO REFINE FREELY: HIS57 HD1, HIS57 HE1, HIS57 HE2, SER214 HG, SER195 HN, GLY193 HN, AND B2V203 H1. DURING THE FINAL STAGES OF REFINEMENT, GEOMETRICAL RESTRAINTS WERE RELEASED FOR ALL NON- HYDROGEN ATOMS IN RESIDUES WITH SINGLE CONFORMATIONS. THE BORON IN RESIDUE B2V WAS REFINED AS A CARBON ATOM, ALLOWING REFINEMENT OF THE OCCUPANCY OF THIS ATOM TO ESTIMATE THE ELECTRON CONTENT (AND NEGATIVE CHARGE) AT THIS LOCATION. TO AID THE READER IN ANALYZING THE STRUCTURE IN THIS PDB FILE, THE ATOM HAS BEEN RE-NAMED TO "B", AND THE OCCUPANCY OF THE BORON ATOM MANUALLY CHANGED TO THE CORRESPONDING OCCUPANCY (1.07; REFINEMENT OF A CARBON IN THIS POSITION RESULTED IN AN OCCUPANCY OF 0.89). SEE PUBLICATION FOR MORE DETAILS.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 21 / Occupancy sum hydrogen: 1367.28 / Occupancy sum non hydrogen: 1784.32 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.9→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
