 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2est: Crystallographic study of the binding of a trifluoroacetyl dipept... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2est | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystallographic study of the binding of a trifluoroacetyl dipeptide anilide inhibitor with elastase | ||||||
 Components Components | ELASTASE | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR COMPLEX / SERINE PROTEINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationpancreatic elastase / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.5 Å X-RAY DIFFRACTION / Resolution: 2.5 Å | ||||||
 Authors Authors | Sieker, L.C. / Hughes, D.L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1982 Title: Crystallographic study of the binding of a trifluoroacetyl dipeptide anilide inhibitor with elastase. Authors: Hughes, D.L. / Sieker, L.C. / Bieth, J. / Dimicoli, J.L. #1: Journal: Eur.J.Biochem. / Year: 1980Title: The Indirect Mechanism of Action of the Trifluoroacetyl Peptides on Elastase Authors: Dimicoli, J.-L. / Renaud, A. / Bieth, J. #2: Journal: J.Mol.Biol. / Year: 1978Title: The Atomic Structure of Crystalline Porcine Pancreatic Elastase at 2.5 Angstroms Resolution. Comparisons with the Structure of Alpha-Chymotrypsin Authors: Sawyer, L. / Shotton, C.M. / Campbell, J.W. / Wendell, P.L. / Muirhead, H. / Watson, H.C. / Diamond, R. / Ladner, R.C. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE TWO SEVEN STRANDED SHEETS IN THIS STRUCTURE ARE REALLY SIX STRANDED BETA BARRELS - THIS ...SHEET THE TWO SEVEN STRANDED SHEETS IN THIS STRUCTURE ARE REALLY SIX STRANDED BETA BARRELS - THIS IS DENOTED BY THE FIRST STRAND RECURRING AS THE LAST STRAND. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2est.cif.gz | 63.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2est.ent.gz | 43.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2est.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/es/2estftp://data.pdbj.org/pub/pdb/validation_reports/es/2est | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: SEE REMARK 7. |
-Components
| #1: Protein | Mass: 25928.031 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-2Z5 / 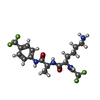  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 457.391 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23F6N4O3 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 457.391 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23F6N4O3References: Trifluoroacetyl dipeptide anilide inhibitor, TFA LYS ALA ANI |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | THE RESIDUE NUMBERING IS BASED ON THAT OF CHYMOTRYPSINOGEN A. THE ENZYME HAS BEEN ASSIGNED CHAIN ...THE RESIDUE NUMBERING IS BASED ON THAT OF CHYMOTRYPS |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 42.85 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 5 / Method: unknown / Details: Shotton, D.M., (1968) J.Mol.Biol., 32, 155. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 2.5 Å / Num. obs: 8481 / Num. measured all: 15401 |
|---|
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor obs: 0.21 / Highest resolution: 2.5 Å Details: NOTE THAT THE SG - SG DISTANCES FOR THE FOUR DISULFIDE BRIDGES ARE ALL ABOUT 3 ANGSTROMS. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2.5 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 7187 / σ(I): 2 / Rfactor obs: 0.21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 15.7 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_angle_d / Dev ideal: 4 |
