 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2e5y: Epsilon subunit and ATP complex of F1F0-ATP synthase from the The... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2e5y | ||||||
|---|---|---|---|---|---|---|---|
| Title | Epsilon subunit and ATP complex of F1F0-ATP synthase from the Thermophilic Bacillus PS3 | ||||||
 Components Components | ATP synthase epsilon chain | ||||||
 Keywords Keywords | HYDROLASE / ATP synthase / F1FO ATP synthase / F1-ATPase / Epsilon subunit / ATP | ||||||
| Function / homology |  Function and homology information Function and homology informationproton-transporting ATP synthase complex / proton-transporting ATP synthase activity, rotational mechanism / ATP binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.92 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.92 Å | ||||||
 Authors Authors | Yagi, H. / Akutsu, H. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2007 Title: Structures of the thermophilic F1-ATPase {varepsilon} subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1 Authors: Yagi, H. / Kajiwara, N. / Tanaka, H. / Tsukihara, T. / Kato-Yamada, Y. / Yoshida, M. / Akutsu, H. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE The 74th residues is LYS and 96th-103th residues are AKERAERR instead of RKSGRTP according ...SEQUENCE The 74th residues is LYS and 96th-103th residues are AKERAERR instead of RKSGRTP according to Kato-Yamada Y., Yoshida M., Hisabori T. [J.Biol.Chem. 275:35746-35750(2000).]. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2e5y.cif.gz | 68.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2e5y.ent.gz | 51.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2e5y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e5/2e5yftp://data.pdbj.org/pub/pdb/validation_reports/e5/2e5y | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2e5tC  2e5uC  1aqtS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 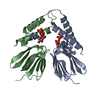
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14585.905 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P07678, H+-transporting two-sector ATPase #2: Chemical |   Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.23 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4 Details: 5% PEG 6000, 0.1M citric acid, pH 4.0, Vapor diffusion, hanging drop, temperature 293.0K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL44XU / Wavelength: 0.9 / Beamline: BL44XU / Wavelength: 0.9 |
| Detector | Type: Bruker DIP-6040 / Detector: CCD / Date: Apr 18, 2004 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1.92→10 Å / Num. obs: 17451 / % possible obs: 92.5 % / Biso Wilson estimate: 27.95 Å2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AQT Resolution: 1.92→10 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.922 / SU B: 3.903 / SU ML: 0.116 / Cross valid method: THROUGHOUT / ESU R: 0.212 / ESU R Free: 0.182
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.821 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.92→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.92→1.968 Å / Total num. of bins used: 20
|
