 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 2cme | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | The crystal structure of SARS coronavirus ORF-9b protein | ||||||
 要素 要素 | (HYPOTHETICAL PROTEIN ...) x 4 | ||||||
 キーワード キーワード | HYPOTHETICAL PROTEIN / ALTERNATIVE OPEN READING FRAME / LIPID-BINDING / VIRUS ASSEMBLY | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報outer mitochondrial membrane protein complex / negative regulation of defense response to virus / positive regulation of autophagosome assembly / Translation of Replicase and Assembly of the Replication Transcription Complex / host cell endoplasmic reticulum / host cell mitochondrion / negative regulation of mitochondrial fission / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / host cell cytoplasmic vesicle membrane / virion component ...outer mitochondrial membrane protein complex / negative regulation of defense response to virus / positive regulation of autophagosome assembly / Translation of Replicase and Assembly of the Replication Transcription Complex / host cell endoplasmic reticulum / host cell mitochondrion / negative regulation of mitochondrial fission / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / host cell cytoplasmic vesicle membrane / virion component / SARS-CoV-1 activates/modulates innate immune responses / mitochondrial outer membrane / symbiont-mediated suppression of host type I interferon-mediated signaling pathway / host cell nucleus / identical protein binding 類似検索 - 分子機能 | ||||||
| 生物種 |   HUMAN SARS CORONAVIRUS (ウイルス) HUMAN SARS CORONAVIRUS (ウイルス) | ||||||
| 手法 |  X線回折 / シンクロトロン / 単波長異常分散 / 解像度: 2.8 Å X線回折 / シンクロトロン / 単波長異常分散 / 解像度: 2.8 Å | ||||||
 データ登録者 データ登録者 | Meier, C. / Aricescu, A.R. / Assenberg, R. / Aplin, R.T. / Gilbert, R.J.C. / Grimes, J.M. / Stuart, D.I. | ||||||
 引用 引用 | ジャーナル: Structure / 年: 2006 タイトル: The Crystal Structure of Orf-9B, a Lipid Binding Protein from the Sars Coronavirus. 著者: Meier, C. / Aricescu, A.R. / Assenberg, R. / Aplin, R.T. / Gilbert, R.J.C. / Grimes, J.M. / Stuart, D.I. | ||||||
| 履歴 |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 2cme.cif.gz | 130 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb2cme.ent.gz | 103.5 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 2cme.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/cm/2cmeftp://data.pdbj.org/pub/pdb/validation_reports/cm/2cme | HTTPS FTP |
|---|
-関連構造データ










-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 単位格子 |
| ||||||||
| 非結晶学的対称性 (NCS) | NCS oper: (Code: given Matrix: (-0.34249, -0.55537, 0.7578), ベクター: |
-要素
-HYPOTHETICAL PROTEIN ... , 4種, 8分子 ABCDFHEG
| #1: タンパク質 | 分子量: 8591.003 Da / 分子数: 1 / 由来タイプ: 組換発現 由来: (組換発現) HUMAN SARS CORONAVIRUS (ウイルス)株: HKU-39849 / 細胞株: VERO E6 / プラスミド: GATEWAY / 発現宿主:  | ||
|---|---|---|---|
| #2: タンパク質 | 分子量: 8748.196 Da / 分子数: 1 / 由来タイプ: 組換発現 詳細: CONTAINS LIPID MOLECULE (MODELLED AS DECANE, RESIDUE NAME D10) 由来: (組換発現) HUMAN SARS CORONAVIRUS (ウイルス)株: HKU-39849 / 細胞株: VERO E6 / プラスミド: GATEWAY / 発現宿主: | ||
| #3: タンパク質 | 分子量: 8420.795 Da / 分子数: 4 / 由来タイプ: 組換発現 詳細: CONTAINS LIPID MOLECULE (MODELLED AS DECANE, RESIDUE NAME D10) 由来: (組換発現) HUMAN SARS CORONAVIRUS (ウイルス)株: HKU-39849 / 細胞株: VERO E6 / プラスミド: GATEWAY / 発現宿主: #4: タンパク質 | 分子量: 8519.924 Da / 分子数: 2 / 由来タイプ: 組換発現 由来: (組換発現) HUMAN SARS CORONAVIRUS (ウイルス)株: HKU-39849 / 細胞株: VERO E6 / プラスミド: GATEWAY / 発現宿主: |
-非ポリマー , 2種, 11分子 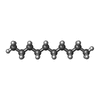

| #5: 化合物 | ChemComp-D10 /  分子量: 142.282 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H22 分子量: 142.282 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H22#6: 水 | ChemComp-HOH / | 分子量: 18.015 Da / 分子数: 7 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.38 Å3/Da / 溶媒含有率: 48 % |
|---|---|
| 結晶化 | pH: 8.2 詳細: 32% PEG3350, 200MM MGCL2, 100MM TRIS-HCL PH8.2, pH 8.20 |
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ESRF  / ビームライン: BM14 / 波長: 0.97903 / ビームライン: BM14 / 波長: 0.97903 |
| 検出器 | タイプ: MARRESEARCH / 検出器: CCD / 日付: 2004年11月9日 / 詳細: MIRRORS |
| 放射 | モノクロメーター: SILICON 111 / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.97903 Å / 相対比: 1 |
| 反射 | 解像度: 2.8→19.9 Å / Num. obs: 22040 / % possible obs: 99.8 % / Observed criterion σ(I): -3 / 冗長度: 14.9 % / Biso Wilson estimate: 84 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 22.8 |
| 反射 シェル | 解像度: 2.8→2.9 Å / 冗長度: 15.1 % / Mean I/σ(I) obs: 1.3 / % possible all: 100 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 単波長異常分散 / 解像度: 2.8→19.9 Å / Data cutoff high absF: 10000 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: RESIDUAL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 80 Å2 / ksol: 0.33 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.8→19.9 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Rms dev Biso : 0.2722 Å2 / Rms dev position: 0.2136 Å / Weight Biso : 3 / Weight position: 40 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.8→2.9 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
