| Entry | Database: PDB / ID: 2ckk
|
|---|
| Title | High resolution crystal structure of the human kin17 C-terminal domain containing a kow motif |
|---|
 Components Components | KIN17 |
|---|
 Keywords Keywords | NUCLEAR PROTEIN / BETA BARREL / RIBOSOMAL PROTEIN / RIBONUCLEOPROTEIN |
|---|
| Function / homology |  Function and homology information Function and homology information
Protein methylation / nuclear matrix / mRNA processing / double-stranded DNA binding / DNA recombination / DNA replication / DNA repair / DNA damage response / protein-containing complex / DNA binding ...Protein methylation / nuclear matrix / mRNA processing / double-stranded DNA binding / DNA recombination / DNA replication / DNA repair / DNA damage response / protein-containing complex / DNA binding / RNA binding / zinc ion binding / nucleoplasm / nucleus / cytosolSimilarity search - Function DNA/RNA-binding protein Kin17, WH-like domain / KIN17-like protein / DNA/RNA-binding protein KIN17, WH-like domain superfamily / KN17, SH3-like C-terminal domain / Kin17, KOW domain / : / KIN17 WH-like domain / KN17 SH3-like domain / KIN17 C-terminal SH3 domain / KIN17 C2H2-type zinc finger domain ...DNA/RNA-binding protein Kin17, WH-like domain / KIN17-like protein / DNA/RNA-binding protein KIN17, WH-like domain superfamily / KN17, SH3-like C-terminal domain / Kin17, KOW domain / : / KIN17 WH-like domain / KN17 SH3-like domain / KIN17 C-terminal SH3 domain / KIN17 C2H2-type zinc finger domain / Domain of Kin17 curved DNA-binding protein / SH3 type barrels. - #30 / SH3 type barrels. - #140 / Zinc finger C2H2 superfamily / Zinc finger C2H2 type domain signature. / SH3 type barrels. / Roll / Ribosomal protein L2, domain 2 / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.45 Å |
|---|
 Authors Authors | le Maire, A. / Schiltz, M. / Pinon-Lataillade, G. / Stura, E. / Couprie, J. / Gondry, M. / Angulo-Mora, J. / Zinn-Justin, S. |
|---|
 Citation Citation | Journal: J.Mol.Biol. / Year: 2006
Title: A Tandem of SH3-Like Domains Participates in RNA Binding in Kin17, a Human Protein Activated in Response to Genotoxics.
Authors: Le Maire, A. / Schiltz, M. / Stura, E.A. / Pinon-Lataillade, G. / Couprie, J. / Moutiez, M. / Gondry, M. / Angulo, J.F. / Zinn-Justin, S. |
|---|
| History | | Deposition | Apr 20, 2006 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Oct 4, 2006 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jan 30, 2019 | Group: Data collection / Experimental preparation / Category: exptl_crystal_grow / Item: _exptl_crystal_grow.method |
|---|
| Revision 1.2 | Feb 6, 2019 | Group: Data collection / Experimental preparation / Category: exptl_crystal_grow / Item: _exptl_crystal_grow.temp |
|---|
| Revision 1.3 | May 8, 2024 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads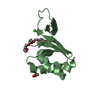









 PDBj
PDBj







 Assembly
Assembly


 Mass: 126.904 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: I
Mass: 126.904 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: I
 Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 18.015 Da / Num. of mol.: 211 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 211 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BM30A / Wavelength: 1.13
/ Beamline: BM30A / Wavelength: 1.13  Processing
Processing