SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 11-STRANDED BARREL THIS IS REPRESENTED BY A 12-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 11-STRANDED BARREL THIS IS REPRESENTED BY A 12-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Mass: 36434.523 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: SCHIFF BASE LINK BETWEEN LYS 200 AND DSB, SCHIFF BASE LINK BETWEEN LYS 253 AND DSB Source: (gene. exp.) CHLOROBIUM VIBRIOFORME (bacteria) / Production host: ESCHERICHIA COLI (E. coli) / References: UniProt: Q59334, porphobilinogen synthase
Resolution: 2.6→89.44 Å / Cor.coef. Fo:Fc: 0.926 / Cor.coef. Fo:Fc free: 0.877 / SU B: 17.322 / SU ML: 0.361 / Cross valid method: THROUGHOUT / ESU R Free: 0.476 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THERE WAS NO ELECTRON DENSITY TO INDICATE THE POSITIONS OF RESIDUES 1 TO 9 SO THESE RESIDUES HAVE BEEN OMITTED FROM THE STRUCTURE.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.32
980
5.2 %
RANDOM
Rwork
0.26
-
-
-
obs
0.263
17930
90 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information CHLOROBIUM VIBRIOFORME (bacteria)
CHLOROBIUM VIBRIOFORME (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg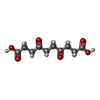
 Mass: 230.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14O6
Mass: 230.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14O6 Mass: 18.015 Da / Num. of mol.: 254 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 254 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing