 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bzi: CRYSTAL STRUCTURE OF THE HUMAN PIM1 IN COMPLEX WITH A RUTHENIUM O... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bzi | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE HUMAN PIM1 IN COMPLEX WITH A RUTHENIUM ORGANOMETALLIC LIGAND RU2 | ||||||
 Components Components | PROTO-ONCOGENE SERINE THREONINE PROTEIN KINASE PIM1 | ||||||
 Keywords Keywords | TRANSFERASE / PIM1 / KINASE / CANCER / LEUKEMIA / NUCLEAR PROTEIN / NUCLEOTIDE-BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / positive regulation of protein serine/threonine kinase activity / ribosomal small subunit binding ...regulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / positive regulation of protein serine/threonine kinase activity / ribosomal small subunit binding / : / positive regulation of cardiac muscle cell proliferation / positive regulation of brown fat cell differentiation / Signaling by FLT3 fusion proteins / positive regulation of TORC1 signaling / regulation of mitotic cell cycle / negative regulation of innate immune response / protein serine/threonine kinase activator activity / cellular response to type II interferon / protein autophosphorylation / manganese ion binding / Interleukin-4 and Interleukin-13 signaling / protein phosphorylation / non-specific serine/threonine protein kinase / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / apoptotic process / negative regulation of apoptotic process / positive regulation of DNA-templated transcription / nucleolus / nucleoplasm / ATP binding / nucleus / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Debreczeni, J.E. / Bullock, A. / Knapp, S. / von Delft, F. / Sundstrom, M. / Arrowsmith, C. / Weigelt, J. / Edwards, A. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of the Human Pim1 in Complex with Ruthenium Organometallic Ligands Authors: Debreczeni, J.E. / Bullock, A. / Knapp, S. / von Delft, F. / Sundstrom, M. / Arrowsmith, C. / Weigelt, J. / Edwards, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bzi.cif.gz | 78.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bzi.ent.gz | 57.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2bzi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bz/2bziftp://data.pdbj.org/pub/pdb/validation_reports/bz/2bzi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2bzhC  2bzjC  1xwsS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



























-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35712.578 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Details: ISOFORM MUTATION R250G / Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: P11-TORONTO / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-DW2 / 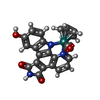  Mass: 496.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H13N3O4Ru Mass: 496.437 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H13N3O4Ru | ||||
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O | ||||
| Compound details | ENGINEERED| Has protein modification | Y | Sequence details | ISOFORM MUTATION R250G | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 60.65 % |
|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop Details: 0.1 M SUCCINATE PHOSPHATE GLYCINE BUFFER PH 6, 30% PEG1K, 0.5% DMSO, SITTING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.2 / Wavelength: 0.981 / Beamline: 14.2 / Wavelength: 0.981 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 4, 2005 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.981 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→50 Å / Num. obs: 34129 / % possible obs: 97.1 % / Observed criterion σ(I): 3 / Redundancy: 5.1 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 15.7 |
| Reflection shell | Resolution: 1.9→2 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.24 / Mean I/σ(I) obs: 3.4 / % possible all: 97 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1XWS Resolution: 1.9→40 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.92 / SU B: 9.881 / SU ML: 0.132 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.146 / ESU R Free: 0.148 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.54 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|