 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2btm: DOES THE HIS12-LYS13 PAIR PLAY A ROLE IN THE ADAPTATION OF THERMO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2btm | ||||||
|---|---|---|---|---|---|---|---|
| Title | DOES THE HIS12-LYS13 PAIR PLAY A ROLE IN THE ADAPTATION OF THERMOPHILIC TIMS TO HIGH TEMPERATURES? | ||||||
 Components Components | PROTEIN (TRIOSEPHOSPHATE ISOMERASE) | ||||||
 Keywords Keywords | ISOMERASE / THERMOPHILIC TRIOSE-PHOSPHATE / GLYCOLYSIS | ||||||
| Function / homology |  Function and homology information Function and homology informationtriose-phosphate isomerase / triose-phosphate isomerase activity / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / gluconeogenesis / glycolytic process / cytosol Similarity search - Function | ||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Delboni, L.F. / Mande, S.C. / Hol, W.G.J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1999 Title: Lys13 plays a crucial role in the functional adaptation of the thermophilic triose-phosphate isomerase from Bacillus stearothermophilus to high temperatures. Authors: Alvarez, M. / Wouters, J. / Maes, D. / Mainfroid, V. / Rentier-Delrue, F. / Wyns, L. / Depiereux, E. / Martial, J.A. #1: Journal: J.Mol.Biol. / Year: 1993 Title: Cloning and overexpression of the triosephosphate isomerase genes from psychrophilic and thermophilic bacteria. Structural comparison of the predicted protein sequences. Authors: Rentier-Delrue, F. / Mande, S.C. / Moyens, S. / Terpstra, P. / Mainfroid, V. / Goraj, K. / Lion, M. / Hol, W.G. / Martial, J.A. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Identification of a potential metal cation-pi binding site in the structure of a thermophilic Bacillus stearothermophilus triosephosphate isomerase mutant. Authors: Wouters, J. / Maes, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2btm.cif.gz | 103.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2btm.ent.gz | 80.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2btm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bt/2btmftp://data.pdbj.org/pub/pdb/validation_reports/bt/2btm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1btmS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26981.641 Da / Num. of mol.: 2 / Mutation: H12N, K13G, P230A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus stearothermophilus (bacteria)Production host: #2: Chemical | 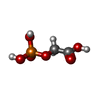  Mass: 156.031 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5O6P Mass: 156.031 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5O6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.62 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 297 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.5 / Wavelength: 1.1 / Beamline: PX9.5 / Wavelength: 1.1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→30 Å / Num. obs: 22732 / % possible obs: 94.3 % / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Rmerge(I) obs: 0.073 / Rsym value: 0.076 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 3 % / Rmerge(I) obs: 0.278 / Rsym value: 0.292 / % possible all: 65 |
| Reflection | *PLUS Num. measured all: 70035 |
| Reflection shell | *PLUS % possible obs: 65 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BTM Resolution: 2.4→30 Å / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE INITIAL STAGES OF REFINEMENT WERE CARRIED OUT WITH X-PLOR
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.44 Å / Total num. of bins used: 22 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.4A / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 30 Å / σ(F): 0 / % reflection Rfree: 5 % / Rfactor Rfree: 0.22 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
