 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ag9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Y137S mutant of GM2-Activator Protein | ||||||
 Components Components | Ganglioside GM2 activator | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / conformational changes in mobile loop (W131 loop) | ||||||
| Function / homology |  Function and homology information Function and homology informationsphingolipid activator protein activity / beta-N-acetylgalactosaminidase activity / glycosphingolipid catabolic process / lipid carrier activity / Glycosphingolipid catabolism / lipid storage / ganglioside catabolic process / oligosaccharide catabolic process / neuromuscular process controlling balance / phospholipase activator activity ...sphingolipid activator protein activity / beta-N-acetylgalactosaminidase activity / glycosphingolipid catabolic process / lipid carrier activity / Glycosphingolipid catabolism / lipid storage / ganglioside catabolic process / oligosaccharide catabolic process / neuromuscular process controlling balance / phospholipase activator activity / lipid transport / lysosomal lumen / cytoplasmic side of plasma membrane / azurophil granule lumen / basolateral plasma membrane / learning or memory / apical plasma membrane / Neutrophil degranulation / extracellular exosome / extracellular region / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Wright, C.S. / Mi, L.Z. / Lee, S. / Rastinejad, F. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2005 Title: Crystal Structure Analysis of Phosphatidylcholine-GM2-Activator Product Complexes: Evidence for Hydrolase Activity. Authors: Wright, C.S. / Mi, L.Z. / Lee, S. / Rastinejad, F. #1: Journal: J.Mol.Biol. / Year: 2000Title: Crystal Structure of Human GM2- Activator Protein with a Novel beta-cup Topology Authors: Wright, C.S. / Li, S.C. / Rastinejad, F. #2: Journal: J.Mol.Biol. / Year: 2003Title: Structure Analysis of Lipid Complexes of GM2-Activator Protein Authors: Wright, C.S. / Zhao, Q. / Rastinejad, F. #3: Journal: J.Mol.Biol. / Year: 2004Title: Evidence for Lipid Packaging in the Crystal Structure of the GM2-Activator Complex with Platelet Activating Factor Authors: Wright, C.S. / Mi, L.Z. / Rastinejad, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ag9.cif.gz | 83.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ag9.ent.gz | 62.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2ag9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ag/2ag9ftp://data.pdbj.org/pub/pdb/validation_reports/ag/2ag9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2af9C  2ag2C  2ag4SC  2agcC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17751.461 Da / Num. of mol.: 2 / Mutation: Y137S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GM2A / Organ: LIVER, BRAIN, NEURONS / Plasmid: pET16b (Novagen) / Species (production host): Escherichia coli / Production host:  #2: Chemical | ChemComp-IPA /   Mass: 60.095 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C3H8O Mass: 60.095 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C3H8O#3: Chemical | ChemComp-MYR / | 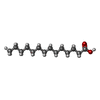  Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 279 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 279 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.04 Å3/Da / Density % sol: 52.1 % |
|---|---|
| Crystal grow | Temperature: 278 K / Method: vapor diffusion, hanging drop / pH: 7.9 Details: Peg 4000, Hepes buffer, isopropanol, pH 7.9, VAPOR DIFFUSION, HANGING DROP, temperature 278K |
-Data collection
| Diffraction | Mean temperature: 123 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 0.92015 Å / Beamline: 22-ID / Wavelength: 0.92015 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jul 30, 2004 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92015 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. all: 28299 / Num. obs: 18838 / % possible obs: 66.6 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 1.7 % / Biso Wilson estimate: 26.7 Å2 / Rmerge(I) obs: 0.082 / Net I/σ(I): 9.9 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 1.1 % / Rmerge(I) obs: 0.291 / Mean I/σ(I) obs: 1.7 / % possible all: 26 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2AG4 Resolution: 2.2→19.87 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 1051432.22 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 72.4487 Å2 / ksol: 0.21949 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→19.87 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.049 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
