 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2aat: 2.8-ANGSTROMS-RESOLUTION CRYSTAL STRUCTURE OF AN ACTIVE-SITE MUTA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2aat | ||||||
|---|---|---|---|---|---|---|---|
| Title | 2.8-ANGSTROMS-RESOLUTION CRYSTAL STRUCTURE OF AN ACTIVE-SITE MUTANT OF ASPARTATE AMINOTRANSFERASE FROM ESCHERICHIA COLI | ||||||
 Components Components | ASPARTATE AMINOTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE(AMINOTRANSFERASE) | ||||||
| Function / homology |  Function and homology information Function and homology information: / L-tyrosine:2-oxoglutarate transaminase activity / L-phenylalanine biosynthetic process / aspartate transaminase / L-aspartate:2-oxoglutarate transaminase activity / pyridoxal phosphate binding / protein homodimerization activity / identical protein binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Smith, D. / Almo, S.C. / Toney, M. / Ringe, D. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1989 Title: 2.8-A-resolution crystal structure of an active-site mutant of aspartate aminotransferase from Escherichia coli. Authors: Smith, D.L. / Almo, S.C. / Toney, M.D. / Ringe, D. #1: Journal: J.Mol.Biol. / Year: 1986Title: Preliminary X-Ray Data for Aspartate Aminotransferase from Escherichia Coli Authors: Smith, D.L. / Ringe, D. / Finlayson, W.L. / Kirsch, J.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2aat.cif.gz | 84.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2aat.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2aat.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aa/2aatftp://data.pdbj.org/pub/pdb/validation_reports/aa/2aat | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES 140 AND 196 ARE CIS PROLINES. | ||||||||
| Details | THE MOLECULE IS A DIMER. TO GENERATE THE OTHER CHAIN ONE MUST APPLY THE CRYSTALLOGRAPHIC SYMMETRY OPERATION (X, 86.74-Y, 79.84-Z) TO THE COORDINATES IN THIS ENTRY. |
-Components
| #1: Protein | Mass: 43561.109 Da / Num. of mol.: 1 / Mutation: K246A Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-PMP / 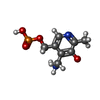  Mass: 248.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H13N2O5P Mass: 248.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H13N2O5P |
| Sequence details | THE RESIDUE NUMBERING USED IN THIS ENTRY HAS BEEN CHOSEN TO MAXIMIZE HOMOLOGY BETWEEN E. COLI ...THE RESIDUE NUMBERING USED IN THIS ENTRY HAS BEEN CHOSEN TO MAXIMIZE HOMOLOGY BETWEEN E. COLI AATASE AND CHICKEN MITOCHONDR |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.11 Å3/Da / Density % sol: 60.51 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 7.5 / Method: batch method | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.8 Å / Num. obs: 8783 / % possible obs: 64 % |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→10 Å /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 10 Å / Num. reflection obs: 8433 / Rfactor obs: 0.22 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 14 Å2 |
