 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 252l | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | GENERATING LIGAND BINDING SITES IN T4 LYSOZYME USING DEFICIENCY-CREATING SUBSTITUTIONS | ||||||
 要素 要素 | T4 LYSOZYME | ||||||
 キーワード キーワード | HYDROLASE / O-GLYCOSYL / T4 LYSOZYME / CAVITY MUTANTS / LIGAND BINDING / PROTEIN ENGINEERING / PROTEIN DESIGN / MULTIPLE CONFORMATIONS | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報viral release from host cell by cytolysis / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / host cell cytoplasm / defense response to bacterium 類似検索 - 分子機能 | ||||||
| 生物種 |  Enterobacteria phage T4 (ファージ) Enterobacteria phage T4 (ファージ) | ||||||
| 手法 |  X線回折 / DIFFERENCE FOURIER / 解像度: 2.1 Å X線回折 / DIFFERENCE FOURIER / 解像度: 2.1 Å | ||||||
 データ登録者 データ登録者 | Baldwin, E.P. / Baase, W.A. / Zhang, X.-J. / Feher, V. / Matthews, B.W. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 1998 タイトル: Generation of ligand binding sites in T4 lysozyme by deficiency-creating substitutions. 著者: Baldwin, E. / Baase, W.A. / Zhang, X. / Feher, V. / Matthews, B.W. #1: ジャーナル: Nature / 年: 1992タイトル: A Cavity-Containing Mutant of T4 Lysozyme is Stabilized by Buried Benzene 著者: Eriksson, A.E. / Baase, W.A. / Wozniak, J.A. / Matthews, B.W. #2: ジャーナル: Methods Enzymol. / 年: 1989タイトル: Expression and Nitrogen-15 Labeling of Proteins for Proton and Nitrogen-15 Nuclear Magnetic Resonance 著者: Muchmore, D.C. / Mcintosh, L.P. / Russell, C.B. / Anderson, D.E. / Dahlquist, F.W. #3: ジャーナル: J.Mol.Biol. / 年: 1987タイトル: Structure of Bacteriophage T4 Lysozyme Refined at 1.7 A Resolution 著者: Weaver, L.H. / Matthews, B.W. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 252l.cif.gz | 50.4 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb252l.ent.gz | 36.1 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 252l.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/52/252lftp://data.pdbj.org/pub/pdb/validation_reports/52/252l | HTTPS FTP |
|---|
-関連構造データ




























-リンク
 PDBj
PDBj




- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 18508.125 Da / 分子数: 1 / 変異: C54T, C97A, M102A, M106A / 由来タイプ: 組換発現 由来: (組換発現) Enterobacteria phage T4 (ファージ)属: T4-like viruses / 生物種: Enterobacteria phage T4 sensu lato / 細胞内の位置: CYTOPLASM / 遺伝子: GENE E / プラスミド: PHS1403 / 遺伝子 (発現宿主): T4 LYSOZYME / 発現宿主:  |
|---|---|
| #2: 化合物 | ChemComp-CL /   分子量: 35.453 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Cl 分子量: 35.453 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Cl |
| #3: 化合物 | ChemComp-HED / 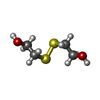  分子量: 154.251 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H10O2S2 分子量: 154.251 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H10O2S2 |
| #4: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 136 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 136 / 由来タイプ: 天然 / 式: H2O |
| 構成要素の詳細 | STRUCTURE OF A T4 LYSOZYME MUTANT THAT CONTAINS TWO DIFFERENT CONFORMATIONS FOR RESIDUES 106 - 114. ...STRUCTURE OF A T4 LYSOZYME MUTANT THAT CONTAINS TWO DIFFERENT CONFORMATI |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.8 Å3/Da / 溶媒含有率: 56.1 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 277 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7 詳細: CRYSTALS GROWN IN HANGING DROPS AT 4 DEGREES C. PROTEIN 10-20 MG/ML IN A BUFFER CONTAINING 0.5 M NACL 0.1 M NAPO4 PH 6.5 WAS DILUTED 1/2 WITH A WELL SOLUTION CONTAINING MIXED K/NAPO4 PH 6.3-7. ...詳細: CRYSTALS GROWN IN HANGING DROPS AT 4 DEGREES C. PROTEIN 10-20 MG/ML IN A BUFFER CONTAINING 0.5 M NACL 0.1 M NAPO4 PH 6.5 WAS DILUTED 1/2 WITH A WELL SOLUTION CONTAINING MIXED K/NAPO4 PH 6.3-7.1,1.8-2.2 MOLAR., pH 7.0, vapor diffusion - hanging drop, temperature 277K PH範囲: 6.3-7.1 | ||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 手法: batch method / 詳細: Remington, S.J., (1978). J. Mol. Biol., 118, 81. / pH: 6.7 | ||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 298 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RUH2R / 波長: 1.5418 |
| 検出器 | タイプ: XUONG-HAMLIN MULTIWIRE / 検出器: AREA DETECTOR / 日付: 1993年3月25日 / 詳細: GRAPHITE MONOCHROMATOR |
| 放射 | モノクロメーター: GRAPHITE(002) / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.1→30 Å / Num. obs: 11176 / % possible obs: 85 % / Rmerge(I) obs: 0.049 |
| 反射 シェル | 最高解像度: 2.1 Å / Rmerge(I) obs: 0.2 / Mean I/σ(I) obs: 2 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: DIFFERENCE FOURIER 開始モデル: CYS-FREE WILD TYPE LYSOZYME WITH RESIDUES 106 - 114 DELETED 解像度: 2.1→30 Å / σ(F): 0 / 立体化学のターゲット値: TNT PROTGEO 詳細: THE STARTING MODEL WAS CYS-FREE WILDTYPE WITH RESIDUES 106 - 115 DELETED AND 102 CONVERTED TO ALA. RESIDUES 106 - 114 WERE FIRST FIT IN A NON-WILDTYPE CONFORMATION. AFTER REFINEMENT THE ...詳細: THE STARTING MODEL WAS CYS-FREE WILDTYPE WITH RESIDUES 106 - 115 DELETED AND 102 CONVERTED TO ALA. RESIDUES 106 - 114 WERE FIRST FIT IN A NON-WILDTYPE CONFORMATION. AFTER REFINEMENT THE WILDTYPE CONFORMATION WAS VISIBLE IN FO-FC DIFFERENCE MAPS, AND WAS ADDED TO THE MODEL. RELATIVE OCCUPANCIES WERE ESTIMATED BASED ON REFINED GROUP OCCUPANCIES (INITIALLY AT 0.5) AND EITHER FIXING OR REFINING THE B VALUES AND POSITIONS. THE WEIGHTS OF EACH CONFORMER ARE 0.6 (ALT) AND 0.4 (WT). IN MOST T4 MUTANT STRUCTURES, RESIDUES 163 AND 164 ARE DISORDERED AND NOT OBSERVED IN ELECTRON DENSITY MAPS. IN THIS MUTANT, THESE RESIDUES WERE RESOLVED IN REFINED FO-FC DIFFERENCE MAPS AND THE RESIDUES ADDED TO THE MODEL.
| ||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: BABINET SCALING / Bsol: 630 Å2 / ksol: 1.1 e/Å3 | ||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.1→30 Å
| ||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: TNT / バージョン: 5F / 分類: refinement | ||||||||||||||||||||||||||||||
| 精密化 | *PLUS Rfactor all: 0.163 | ||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
