 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1zzj: Structure of the third KH domain of hnRNP K in complex with 15-me... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1zzj | ||||||
|---|---|---|---|---|---|---|---|
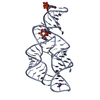
| Title | Structure of the third KH domain of hnRNP K in complex with 15-mer ssDNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | STRUCTURAL PROTEIN/DNA / PROTEIN-DNA COMPLEX / STRUCTURAL PROTEIN-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator / positive regulation of low-density lipoprotein particle clearance / random inactivation of X chromosome / regulatory ncRNA-mediated heterochromatin formation / regulation of mRNA splicing, via spliceosome / SUMOylation of RNA binding proteins / podosome / Processing of Capped Intron-Containing Pre-mRNA / negative regulation of mRNA splicing, via spliceosome / RNA processing ...regulation of intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator / positive regulation of low-density lipoprotein particle clearance / random inactivation of X chromosome / regulatory ncRNA-mediated heterochromatin formation / regulation of mRNA splicing, via spliceosome / SUMOylation of RNA binding proteins / podosome / Processing of Capped Intron-Containing Pre-mRNA / negative regulation of mRNA splicing, via spliceosome / RNA processing / catalytic step 2 spliceosome / mRNA Splicing - Major Pathway / HCMV Late Events / mRNA splicing, via spliceosome / cytoplasmic stress granule / cadherin binding / ribonucleoprotein complex / protein domain specific binding / focal adhesion / negative regulation of DNA-templated transcription / mRNA binding / regulation of transcription by RNA polymerase II / negative regulation of apoptotic process / chromatin / signal transduction / positive regulation of transcription by RNA polymerase II / DNA binding / RNA binding / extracellular exosome / nucleoplasm / membrane / identical protein binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Backe, P.H. / Messias, A.C. / Ravelli, R.B. / Sattler, M. / Cusack, S. | ||||||
 Citation Citation | Journal: STRUCTURE / Year: 2005 Title: X-Ray Crystallographic and NMR Studies of the Third KH Domain of hnRNP K in Complex with Single-Stranded Nucleic Acids Authors: Backe, P.H. / Messias, A.C. / Ravelli, R.B. / Sattler, M. / Cusack, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1zzj.cif.gz | 59.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1zzj.ent.gz | 43.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1zzj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zz/1zzjftp://data.pdbj.org/pub/pdb/validation_reports/zz/1zzj | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj










































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1
|