S. PYOGENES IMPDH IS A TETRAMER WITH ITS FOUR SUBUNITS RELATED BY A CRYSTALLOGRAPHIC FOURFOLD AXIS. EACH MONOMER HAS A TWO-DOMAIN STRUCTURE: A CATALYTIC DOMAIN (AMINO ACID RESIDUES 2-92 AND 224-492) FORMING THE INTERIOR CORE OF THE ACTIVE TETRAMERIC ENZYME AND A CBS DIMER DOMAIN (RESIDUES 93-223) PROJECTING OUTWARD FROM THE CORNERS OF THE SQUARE. THE CBS DESIGNATION ARISES FROM THE ORIGINAL IDENTIFICATION OF THIS FOLDING MOTIF IN THE ENZYME CYSTATHIONINE-"BETA"-SYNTHASE [BATEMAN, A. (1997) TRENDS BIOCHEM. SCI. 22, 12-13]. THE CBS DIMER DOMAIN, FOUND IN IMPDH PROTEINS FROM ALL THREE KINGDOMS, IS COMPOSED OF TWO CBS MOTIFS RELATED BY APPROXIMATE TWOFOLD SYMMETRY (RMS DEVIATIONS BETWEEN ALPHA CARBON ATOMS: 2.7 ANGSTROMS). EACH CBS MOTIF HAS THE CHARACTERISTIC SHEET/HELIX/SHEET/SHEET/HELIX TOPOLOGY. THIS IS THE FIRST REPORTED COMPLETE STRUCTURE OF A CBS DIMER DOMAIN, A FOLDING MOTIF PROPOSED TO ACT AS A REGULATORY ELEMENT SINCE MUTATIONS LEAD TO THE HUMAN DISEASE HOMOCYSTINURIA. EACH IPMDH MONOMER CONTAINS IMP IN THE CATALYTIC SITE. THIS SUBSTRATE IS NOT COVALENTLY BOUND TO THE ACTIVE SITE CYS310 SUGGESTING THAT IMP DOES NOT FORM A COVALENT BOND IN THE ABSENCE OF NAD.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Streptococcus pyogenes (化膿レンサ球菌)
Streptococcus pyogenes (化膿レンサ球菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード






 PDBj
PDBj
 集合体
集合体


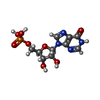
 分子量: 348.206 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H13N4O8P
分子量: 348.206 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H13N4O8P 分子量: 18.015 Da / 分子数: 499 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 499 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 19-ID / 波長: 0.9791,1.0781
/ ビームライン: 19-ID / 波長: 0.9791,1.0781 解析
解析