 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3tsd: Crystal Structure of Inosine-5'-monophosphate Dehydrogenase from ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3tsd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Inosine-5'-monophosphate Dehydrogenase from Bacillus anthracis str. Ames complexed with XMP | |||||||||
 Components Components | Inosine-5'-monophosphate dehydrogenase | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / TIM barrel / CBS domain / cytosol / Structural Genomics / Center for Structural Genomics of Infectious Diseases / CSGID | |||||||||
| Function / homology |  Function and homology information Function and homology informationIMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / GTP biosynthetic process / nucleotide binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.653 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.653 Å | |||||||||
 Authors Authors | Kim, Y. / Makowska-Grzyska, M. / Hasseman, J. / Anderson, W.F. / Joachimiak, A. / Center for Structural Genomics of Infectious Diseases (CSGID) | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2012 Title: Bacillus anthracis inosine 5'-monophosphate dehydrogenase in action: the first bacterial series of structures of phosphate ion-, substrate-, and product-bound complexes. Authors: Makowska-Grzyska, M. / Kim, Y. / Wu, R. / Wilton, R. / Gollapalli, D.R. / Wang, X.K. / Zhang, R. / Jedrzejczak, R. / Mack, J.C. / Maltseva, N. / Mulligan, R. / Binkowski, T.A. / Gornicki, P. ...Authors: Makowska-Grzyska, M. / Kim, Y. / Wu, R. / Wilton, R. / Gollapalli, D.R. / Wang, X.K. / Zhang, R. / Jedrzejczak, R. / Mack, J.C. / Maltseva, N. / Mulligan, R. / Binkowski, T.A. / Gornicki, P. / Kuhn, M.L. / Anderson, W.F. / Hedstrom, L. / Joachimiak, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3tsd.cif.gz | 347.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3tsd.ent.gz | 282.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3tsd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ts/3tsdftp://data.pdbj.org/pub/pdb/validation_reports/ts/3tsd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3tsbC  3usbC  1zfjS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | tetramer is formed from either chain A or chain B by applying x,y,z; -y,x,z; y,-x,z; -x,-y,z |
-Components
| #1: Protein | Mass: 55183.172 Da / Num. of mol.: 2 / Fragment: IMPDH / Mutation: full-length Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: Q81W29, UniProt: A0A6L8P2U9*PLUS, IMP dehydrogenase #2: Chemical | 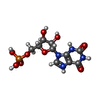  Mass: 365.213 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N4O9P Mass: 365.213 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N4O9P#3: Chemical | ChemComp-TAR / | 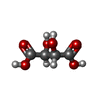  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6#4: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 95 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 95 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.52 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.8 M sodium/potassium tartrate tetrahydrate, 0.1 M Tris pH8.5, 0.5 % PEGMME 5000, VAPOR DIFFUSION, SITTING DROP, temperature 289K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97918 Å / Beamline: 19-ID / Wavelength: 0.97918 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Mar 26, 2011 / Details: mirrors |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97918 Å / Relative weight: 1 |
| Reflection | Resolution: 2.65→50 Å / Num. all: 30553 / Num. obs: 30553 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 9.1 % / Biso Wilson estimate: 56.56 Å2 / Rsym value: 0.094 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 2.65→2.7 Å / Redundancy: 9.2 % / Mean I/σ(I) obs: 3.2 / Num. unique all: 1502 / Rsym value: 0.735 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ID 1ZFJ monomer Resolution: 2.653→38.975 Å / SU ML: 0.6 / Isotropic thermal model: mixed / Cross valid method: THROUGHOUT / σ(F): 0 / Phase error: 25.81 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.73 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 48.99 Å2 / ksol: 0.307 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 82.7 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.653→38.975 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
