 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ywm: Crystal structure of the N-terminal domain of group B Streptococc... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ywm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the N-terminal domain of group B Streptococcus alpha C protein | ||||||
 Components Components | C protein alpha-antigen | ||||||
 Keywords Keywords | SURFACE ACTIVE PROTEIN / BETA SANDWICH / FIBRONECTIN FOLD / ANTIPARALLEL THREE-HELIX BUNDLE | ||||||
| Function / homology |  Function and homology information Function and homology informationSurface Active Protein / Surface Active Protein fold / Surface Active Protein domain / Alpha C protein, N-terminal / AlphaC, C-terminal / ACP, C-terminal domain superfamily / ACP, N-terminal domain superfamily / Alpha C protein N terminal / AlphaC N-terminal domain 2 / : ...Surface Active Protein / Surface Active Protein fold / Surface Active Protein domain / Alpha C protein, N-terminal / AlphaC, C-terminal / ACP, C-terminal domain superfamily / ACP, N-terminal domain superfamily / Alpha C protein N terminal / AlphaC N-terminal domain 2 / : / Rib/alpha/Esp surface antigen / Rib domain / M protein-type anchor domain / YSIRK type signal peptide / Substrate Binding Domain Of Dnak; Chain:A; Domain 2 / YSIRK Gram-positive signal peptide / LPXTG cell wall anchor motif / Gram-positive cocci surface proteins LPxTG motif profile. / LPXTG cell wall anchor domain / Up-down Bundle / Sandwich / Mainly Beta / Mainly Alpha Similarity search - Domain/homology | ||||||
| Biological species |  Streptococcus agalactiae (bacteria) Streptococcus agalactiae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.86 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.86 Å | ||||||
 Authors Authors | Auperin, T.C. / Bolduc, G.R. / Baron, M.J. / Heroux, A. / Filman, D.J. / Madoff, L.C. / Hogle, J.M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2005 Title: Crystal structure of the N-terminal domain of the group B streptococcus alpha C protein. Authors: Auperin, T.C. / Bolduc, G.R. / Baron, M.J. / Heroux, A. / Filman, D.J. / Madoff, L.C. / Hogle, J.M. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1992 Title: Large, Identical, Tandem Repeating Units in the C Protein Alpha Antigen Gene, Bca, of Group B Streptococci Authors: Michel, J.L. / Madoff, L.C. / Olson, K. / Kling, D.E. / Kasper, D.L. / Ausubel, F.M. #2: Journal: Cell.Microbiol. / Year: 2002 Title: The Alpha C Protein Mediates Internalization of Group B Streptococcus within Human Cervical Epithelial Cells Authors: Bolduc, G.R. / Baron, M.J. / Gravekamp, C. / Lachenauer, C.S. / Madoff, L.C. #3: Journal: J.Biol.Chem. / Year: 2004 Title: Alpha C Protein of Group B Streptococcus Binds Host Cell Surface Glycosaminoglycan and Enters Cells by an Actin-Dependent Mechanism Authors: Baron, M.J. / Bolduc, G.R. / Goldberg, M.B. / Auperin, T.C. / Madoff, L.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ywm.cif.gz | 55.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ywm.ent.gz | 39.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ywm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yw/1ywmftp://data.pdbj.org/pub/pdb/validation_reports/yw/1ywm | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22022.682 Da / Num. of mol.: 1 / Fragment: N-TERMINAL DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus agalactiae (bacteria) / Strain: A909 / Gene: bca / Plasmid: PET24A / Species (production host): Escherichia coli / Production host: | ||
|---|---|---|---|
| #2: Chemical | ChemComp-DTU / (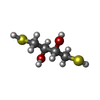  Mass: 154.251 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2S2 Mass: 154.251 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2S2 | ||
| #3: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 57 % Description: The statistics for the Mutant SeMet used for experimental phasing are: resolution range 2.6-30, 97.4% completeness, 8581 reflections, 12.8 redundancy, 6.8 Rsym, 30.7 / Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 4.6 | Details: sodium acetate, PEG4000, DTT, pH 4.60, VAPOR DIFFUSION, HANGING DROP, temperature 294K |
|---|
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation |
| |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 1.86→40 Å / Num. obs: 19465 / % possible obs: 95.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 14 % / Rsym value: 0.053 / Net I/σ(I): 29.88 | |||||||||||||||
| Reflection shell | Resolution: 1.86→1.93 Å / Mean I/σ(I) obs: 4.69 / Rsym value: 0.339 / % possible all: 71.7 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD Starting model: ALPHA C SELENOMETHIONYL MUTANT (2 SELENIUM ATOMS) Resolution: 1.86→39.53 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.924 / SU B: 2.673 / SU ML: 0.08 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.13 / ESU R Free: 0.131 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: THERE IS NO SINGLE WELL ORDERED CONFORMATION FOR LEU227. THE REFINEMENT STATISTICS FOR THE MUTANT SEMET SET ARE RESOLUTION RANGE 2.6-12, 0.269 RWORK, 0.308 RFREE, 0.010 RMSD BOND LENGTH AND ...Details: THERE IS NO SINGLE WELL ORDERED CONFORMATION FOR LEU227. THE REFINEMENT STATISTICS FOR THE MUTANT SEMET SET ARE RESOLUTION RANGE 2.6-12, 0.269 RWORK, 0.308 RFREE, 0.010 RMSD BOND LENGTH AND 1.552 RMSD BOND ANGLE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.69 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.86→39.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.86→1.96 Å / Total num. of bins used: 10 /
|
