 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1y1a | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF CALCIUM AND INTEGRIN BINDING PROTEIN | ||||||
 Components Components | Calcium and integrin binding 1 (calmyrin) | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / CALCIUM-BINDING PROTEIN / INTEGRIN / EF-HAND / GLUTATHIONE / GLUTATHIOLATION | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of male germ cell proliferation / calcium-dependent protein kinase inhibitor activity / endomitotic cell cycle / positive regulation of catalytic activity / filopodium tip / thrombopoietin-mediated signaling pathway / negative regulation of microtubule depolymerization / positive regulation of calcineurin-NFAT signaling cascade / positive regulation of cell adhesion mediated by integrin / platelet formation ...positive regulation of male germ cell proliferation / calcium-dependent protein kinase inhibitor activity / endomitotic cell cycle / positive regulation of catalytic activity / filopodium tip / thrombopoietin-mediated signaling pathway / negative regulation of microtubule depolymerization / positive regulation of calcineurin-NFAT signaling cascade / positive regulation of cell adhesion mediated by integrin / platelet formation / positive regulation of cell migration involved in sprouting angiogenesis / positive regulation of cell-matrix adhesion / negative regulation of protein phosphorylation / positive regulation of protein serine/threonine kinase activity / spermatid development / regulation of cell division / positive regulation of protein targeting to membrane / protein serine/threonine kinase inhibitor activity / negative regulation of megakaryocyte differentiation / extrinsic apoptotic signaling pathway / cytoplasmic microtubule organization / protein-membrane adaptor activity / positive regulation of substrate adhesion-dependent cell spreading / response to ischemia / negative regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / : / cell periphery / positive regulation of protein localization to plasma membrane / cellular response to nerve growth factor stimulus / sarcolemma / positive regulation of protein phosphorylation / cellular response to growth factor stimulus / cellular response to tumor necrosis factor / small GTPase binding / ruffle membrane / double-strand break repair / regulation of cell population proliferation / lamellipodium / negative regulation of neuron projection development / growth cone / positive regulation of cell growth / angiogenesis / vesicle / transmembrane transporter binding / perikaryon / positive regulation of ERK1 and ERK2 cascade / cell adhesion / apical plasma membrane / neuron projection / nuclear body / positive regulation of cell migration / negative regulation of cell population proliferation / axon / cell division / neuronal cell body / apoptotic process / calcium ion binding / positive regulation of cell population proliferation / DNA damage response / centrosome / negative regulation of apoptotic process / perinuclear region of cytoplasm / magnesium ion binding / Golgi apparatus / endoplasmic reticulum / extracellular exosome / nucleoplasm / membrane / nucleus / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å | ||||||
 Authors Authors | Blamey, C.J. / Ceccarelli, C. / Naik, U.P. / Bahnson, B.J. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2005 Title: The crystal structure of calcium- and integrin-binding protein 1: Insights into redox regulated functions Authors: Blamey, C.J. / Ceccarelli, C. / Naik, U.P. / Bahnson, B.J. | ||||||
| History |
| ||||||
| Remark 295 | NON-CRYSTALLOGRAPHIC SYMMETRY THE TRANSFORMATIONS PRESENTED ON THE MTRIX RECORDS BELOW DESCRIBE ... NON-CRYSTALLOGRAPHIC SYMMETRY THE TRANSFORMATIONS PRESENTED ON THE MTRIX RECORDS BELOW DESCRIBE NON-CRYSTALLOGRAPHIC RELATIONSHIPS AMONG ATOMS IN THIS ENTRY. APPLYING THE APPROPRIATE MTRIX TRANSFORMATION TO THE RESIDUES LISTED FIRST WILL YIELD APPROXIMATE COORDINATES FOR THE RESIDUES LISTED SECOND. CHAIN IDENTIFIERS GIVEN AS "?" REFER TO CHAINS FOR WHICH ATOMS ARE NOT FOUND IN THIS ENTRY. APPLIED TO TRANSFORMED TO TRANSFORM CHAIN RESIDUES CHAIN RESIDUES RMSD SSS M 1 A 52 .. 191 B 52 .. 191 0.655 WHERE SSS -> COLUMNS 8-10 OF MTRIX RECORDS | ||||||
| Remark 999 | SEQUENCE GB 12654075 AAH00846 1 - 8 NOT IN ATOMS LIST A DELETION MUTANT OF CIB WHOSE SEQUENCE DOES ...SEQUENCE GB 12654075 AAH00846 1 - 8 NOT IN ATOMS LIST A DELETION MUTANT OF CIB WHOSE SEQUENCE DOES NOT INCLUDE THE FIRST EIGHT N-TERMINAL RESIDUES WAS USED FOR THE X-RAY STUDIES DESCRIBED IN THIS ENTRY. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1y1a.cif.gz | 95.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1y1a.ent.gz | 72.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1y1a.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y1/1y1aftp://data.pdbj.org/pub/pdb/validation_reports/y1/1y1a | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| 4 |
| ||||||||||||
| 5 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 20980.490 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CIB1 / Species (production host): Escherichia coli / Production host:  #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-GSH / | 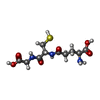  Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S Mass: 307.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N3O6S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 278 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 278 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 6.2 Å3/Da / Density % sol: 80.1 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 20 MG/ML PROTEIN, 50MM HEPES, 3M FORMATE, 300MM NaCl, 1% DMSO, 0.25MM DTT, pH 7.00, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-ID-B / Wavelength: 1.12709 / Wavelength: 1.53578, 1.53466, 1.47954 / Beamline: 14-ID-B / Wavelength: 1.12709 / Wavelength: 1.53578, 1.53466, 1.47954 | |||||||||||||||
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Aug 20, 2004 | |||||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2.3→50 Å / Num. all: 44410 / Num. obs: 44410 / % possible obs: 92.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 10.4 % / Biso Wilson estimate: 45.8 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 7.9 | |||||||||||||||
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 10.1 % / Rmerge(I) obs: 0.388 / Mean I/σ(I) obs: 2 / % possible all: 65.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.3→47.25 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 2357501.5 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 50.7122 Å2 / ksol: 0.353729 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 55.6 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→47.25 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.43 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
