IN THIS ENTRY THE LAST COLUMN REPRESENTS THE AVERAGE RMS DIFFERENCE BETWEEN THE INDIVIDUAL ... IN THIS ENTRY THE LAST COLUMN REPRESENTS THE AVERAGE RMS DIFFERENCE BETWEEN THE INDIVIDUAL SIMULATED ANNEALING STRUCTURES AND THE MEAN COORDINATE POSITIONS. IT IS IMPORTANT TO NOTE THAT THE VALUES GIVEN FOR THE BACKBONE ATOMS AND NON-INTERFACIAL SIDECHAINS PROVIDE ONLY A MEASURE OF THE PRECISION WITH WHICH THE RELATIVE ORIENTATION OF THE TWO PROTEINS HAVE BEEN DETERMINED AND DOES NOT TAKE INTO ACCOUNT THE ERRORS IN THE X-RAY COORDINATES OF HPR AND IIAMAN. RESIDUE NUMBERING: IIAMAN: 1-136 (THE N-TERMINAL METHIONINE IS CLEAVED AND RESIDUES 131-136 WERE DISORDERED. RESIDUES 134-136 ARE CLONING ARTIFACTS; SEE REMARKS IN THE 1PDO CRYSTAL STRUCTURE.) HPR: 201-285 (CORRESPONDING TO RESIDUES 1-85). PHOSPHATES: RESIDUES 401 AND 402. TWO SETS OF COORDINATES ARE GIVEN: MODEL 1: RESTRAINED REGULARIZED MEAN COORDINATES FOR THE MODEL OF THE ASSOCIATIVE PHOSPHORYL TRANSITION STATE HPR-IIAMTL COMPLEX. EXPERIMENTAL RESTRAINTS ARE IDENTICAL TO THOSE USED FOR MODEL 2, BUT COVALENT GEOMETRY RESTRAINTS ARE INCLUDED RELATING TO THE PENTACOORDINATE PHOSPHORYL GROUP IN A TRIGONAL BIPYRAMIDAL GEOMETRY. THE STRUCTURE IS DERIVED FROM MODEL 2 BY RESTRAINED REGULARIZATION IN WHICH ONLY THE ACTIVE SITE HISTIDINES, THE BACKBONE IMMIEDIATELY ADJACENT (ONE RESIDUE ON EITHER SIDE) TO THE ACTIVE SITE HISTIDINES, AND THE INTERFACIAL SIDECHAINS ARE ALLOWED TO MOVE. THE N-P BOND LENGTHS ARE RESTRAINED TO 2 A. IIAMAN-HPR COMPLEX RMS DEVIATIONS FROM NOE DISTANCE RESTRAINTS: 0.009 A RMS DEVIATIONS FROM SIDECHAIN TORSION ANGLE RESTRAINTS: 0.0 DEG. DIPOLAR COUPLING R-FACTORS (CLORE AND GARRETT (1999) J. AM. CHEM. SOC. 121, 9008-9012): IIAMAN HPr NH 11.6 18.5/12.4 MODEL 2: RESTRAINED REGULARIZED MEAN COORDINATES OF THE UNPHOSPHORYLATED IIAMAN-HPR COMPLEX SOLVED ON THE BASIS OF 58x2 INTERMOLECULAR INTERPROTON DISTANCE DISTANCE RESTRAINTS BETWEEN THE TWO MOLECULES OF HPR AND THE IIAMAN DIMER, 47x2 INTRA AND 16x2 INTER-SUBUNIT IIAMAN DISTANCE RESTRAINTS RELATING ONLY TO INTERFACIAL SIDECHAINS, 39x2 INTRAMOLECULAR HPR cwDISTANCE RESTRAINTS RELATING ONLY TO INTERFACIAL SIDECHAINS, 29x2 INTERFACIAL SIDECHAIN TORSION ANGLE RESTRAINTS, 92X2 RESIDUAL DIPOLAR COUPLINGS FOR IIAMAN AND 66X2 RESIDUAL DIPOLAR COUPLINGS FOR HPR. WAS USED FOR THE DIPOLAR COUPLINGS (CLORE AND GARRETT (1999) J. AM. CHEM. SOC. 121, 9008-9012). NOTE THE NH DIPOLAR COUPLINGS FOR FULLY BOUND HPR (I.E. BOTH BIDNING SITES ON IIAMAN FULLY SATURATED) ARE BACKCALCULATED FROM THE DIPOLAR COUPLINGS MEASURED FOR A SAMPLE WITH A THREE-FOLD MOLAR EXCESS OF HPR OVER IIAMAN BINDING SITES AND A SAMPLE OF FREE HPR. THE FIRST VALUE OF THE R-FACTOR USES DIPOLAR COUPLINGS FOR THE FULLY BOUND STATE BACK-CALCULATED USING THE MEASURED VALUES OF THE DIPOLAR OUPLINGS FOR FREE HPR; WHILE THE SECOND NUMBER USES THE CALCULATED VALUES OF THE DIPOLAR COUPLINGS FOR FREE HPR DERIVED FROM THE MEASURED VALUES AND THE CRYSTAL STRUCTURE OF FREE HPR USING SINGULAR VALUE DECOMPOSITION. NOTE A SINGLE ALIGNMENT TENSOR IS USED. THE DIPOLAR COUPLING R-FACTOR OBTAINED BY SINGULAR VALUE DECOMPOSITION AGAINST THE CRYSTAL STRUCTURES OF IIAMAN AND HPR INDIVIDUAL ARE THE SAME AS THOSE OBTAINED FOR THE COMPLEX AS A WHOLE.
(4) TRSOE-BASED 2D, 3D EXPERIMENTS FOR DIPOLAR COUPLINGS. DIPOLAR COUPLINGS WERE MEASURED IN A NEMATIC PHASE OF A 5% PEG/HEXANOL (SURFACTANT TO ALCOHOL RATION OF 0.96)
-
Sample preparation
Sample conditions
Ionic strength: 40 mM SODIUM PHOSPHATE / pH: 6.5 / Temperature: 308.00 K
-
NMR measurement
NMR spectrometer
Type
Manufacturer
Model
Field strength (MHz)
Spectrometer-ID
Bruker AVANCE DMX
Bruker
AVANCEDMX
500
1
Bruker AVANCE DMX
Bruker
AVANCEDMX
600
2
Bruker AVANCE DRX
Bruker
AVANCEDRX
750
3
Bruker AVANCE DRX
Bruker
AVANCEDRX
800
4
Bruker AVANCE DRX
Bruker
AVANCEDRX
800
5
-
Processing
NMR software
Name
Version
Developer
Classification
X-PLOR NIH
(HTTP://NMR.CIT.NIH.GOV/XPLOR_NIH)
CLORE, KUSZEWSKI, SCHWIETERS, TJANDRA
refinement
X-PLOR NIH
CLORE, KUSZEWSKI, SCHWIETERS, TJANDRA
structuresolution
Refinement
Method: CONJOINED RIGID BODY, TORSION ANGLE DYNAMICS / Software ordinal: 1 Details: THE STRUCTURES WERE CALCULATED BY CONJOINED RIGID BODY/TORSION ANGLE DYNAMICS (SCHWIETERS & CLORE (2001) J.MAGN.RESON 152, 288-302). THE TARGET FUNCTIONS COMPRISES TERMS FOR NOE RESTRAINTS, ...Details: THE STRUCTURES WERE CALCULATED BY CONJOINED RIGID BODY/TORSION ANGLE DYNAMICS (SCHWIETERS & CLORE (2001) J.MAGN.RESON 152, 288-302). THE TARGET FUNCTIONS COMPRISES TERMS FOR NOE RESTRAINTS, SIDECHAIN TORSION ANGLE RESTRAINTS, RESIDUAL DIPOLAR COUPLING RESTRAINTS (CLORE ET AL. J.MAGN.RESON. 131, 159-162 (1998); J.MAGN.RESON.133, 216-221(1998)), A RADIUS OF GYRATION TERM (KUSZEWSKI ET AL.(1999), A QUARTIC VAN DER WAALS REPULSION TERM (NILGES ET AL. (1988) FEBS LETT. 229, 129- 136), AND A TORSION ANGLE CONFORMATIONAL DATABASE POTENTIAL OF MEAN FORCE (CLORE AND KUSZEWSKI 2002) J.AM.CHEM.SOC 124, 2866-2867). THE STARTING COORDINATE COME FROM THE X-RAY STRUCTURES (WITH PROTONS ADDED) OF E. COLI HPR (1POH, JIA ET AL. (1993) J.BIOL.CHEM. 268, 22940-22501, RESOLUTION 2.0 A); AND IIAMAN (1PDO, NUNN ET AL. (1996) J.MOL.BI J.MOL.BIOL. 259, 502-511; RESOLUTION 1.7A). THE BACKBONE COORDINATES AND NON-INTERFACIAL SIDECHAINS ARE TREATED AS RIGID BODIES THROUGHOUT WITH THE IIAMAN DIMER HELD FIXED, THE TWO HPR MOLECULES ALLOWED TO ROTATE AND TRANSLATE, AND THE AXIS OF THE SINGLE DIPOLAR COUPLING ALIGNMENT TENSOR FREE TO ROTATE. THE INTERFACIAL SIDECHAINS ARE GIVEN FULL TORSIONAL DEGREES OF FREEDOM.
NMR ensemble
Conformer selection criteria: REGULARIZED MEAN STRUCTURES / Conformers calculated total number: 100 / Conformers submitted total number: 2
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj Assembly
Assembly
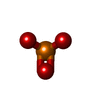
 Mass: 78.972 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO3
Mass: 78.972 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO3 Sample preparation
Sample preparation Processing
Processing X-PLOR NIH
X-PLOR NIH