SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
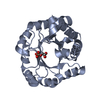
Mass: 25888.586 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, RESIDUES 31-254 Source method: isolated from a genetically manipulated source Details: THE CRYSTAL STRUCTURE IS A COMPLEX WITH A SOAKED CELLOPENTAOSE. THE FIFTH GLUCOSE UNIT IS, HOWEVER, NOT VISIBLE IN THE ELECTRON DENSITY Source: (gene. exp.) HUMICOLA GRISEA (fungus) / Production host: ASPERGILLUS NIGER (mold) / References: UniProt: Q8NJY3, cellulase
Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 1.8 Å3/Da / Density % sol: 30.6 % Description: INITIAL MODEL WAS PRODUCED BY RIGID BODY REFINEMENT USING THE APO PROTEIN STRUCTURE PDB ENTRY 1OLR
Crystal grow
pH: 3.1 Details: CRYSTALS GREW FROM A PROTEIN STOCK SOLUTION CONTAINING 1MG/ML PROTEIN IN 0.05 M BIS TRIS PROPANE AND 0.05 M AMMONIUM ACETATE, PH 8. CRYSTALS WERE CRYOPROTECTED IN UNBUFFERED 50% MME PEG 2000
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.934 Å / Relative weight: 1
Reflection
Resolution: 1.4→42.258 Å / Num. obs: 41450 / % possible obs: 99.7 % / Redundancy: 9.7 % / Rmerge(I) obs: 0.088 / Net I/σ(I): 20.4
Reflection shell
Resolution: 1.4→1.42 Å / Rmerge(I) obs: 0.316 / Mean I/σ(I) obs: 13.8 / % possible all: 99.2
-
Processing
Software
Name
Version
Classification
REFMAC
5.1.24
refinement
DENZO
datareduction
SCALEPACK
datascaling
Refinement
Method to determine structure: OTHER / Resolution: 1.4→42.26 Å / Cor.coef. Fo:Fc: 0.969 / Cor.coef. Fo:Fc free: 0.964 / Cross valid method: THROUGHOUT / ESU R: 0.054 / ESU R Free: 0.055 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FOLLOWING PROTEIN RESIDUES HAVE BEEN MODELED IN MULTIPLE CONFORMATIONS: A49 A141 A167 A174 A183 A224. THE FOLLOWING WATERS HAVE BEEN ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FOLLOWING PROTEIN RESIDUES HAVE BEEN MODELED IN MULTIPLE CONFORMATIONS: A49 A141 A167 A174 A183 A224. THE FOLLOWING WATERS HAVE BEEN MODELED IN MULTIPLE CONFORMATIONS: W17 W40 W45 W73 W75 W78 W112 W113 W114 W127 W147 W170 W171 W180 W186 ATOMS WITH MISSING ELECTRON DENSITY ARE ASSIGNED ZERO OCCUPANCY. ATOMS ARE ASSIGNED REDUCED OCCUPANCIES WHEN ELECTRON DENSITY IS WEAK OR ATOMS HAVE PARTIAL OCCUPANCY.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.168
1298
3.1 %
RANDOM
Rwork
0.147
-
-
-
obs
0.148
40151
100 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HUMICOLA GRISEA (fungus)
HUMICOLA GRISEA (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj
 Assembly
Assembly
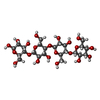


 Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM
Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.934
/ Beamline: ID14-1 / Wavelength: 0.934  Processing
Processing