 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1u4g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Elastase of Pseudomonas aeruginosa with an inhibitor | ||||||
 Components Components | Elastase | ||||||
 Keywords Keywords | HYDROLASE / x-ray structure / Elastase / inhibition / peptidase family M4 | ||||||
| Function / homology |  Function and homology information Function and homology informationpseudolysin / protein transport by the Sec complex / protein secretion by the type II secretion system / single-species biofilm formation / bacterial-type flagellum-dependent swarming motility / metalloendopeptidase activity / endopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.4 Å | ||||||
 Authors Authors | Bitto, E. / McKay, D.B. | ||||||
 Citation Citation | Journal: To be Published / Year: 2004 Title: Elastase of Pseudomonas aeruginosa with an inhibitor Authors: Bitto, E. / McKay, D.B. #1: Journal: J.Biol.Chem. / Year: 1991Title: Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution Authors: Thayer, M.M. / Flaherty, K.M. / McKay, D.B. #2: Journal: Biochemistry / Year: 1992Title: Structural comparison suggests that thermolysin and related neutral proteases undergo hinge-bending motion during catalysis Authors: Holland, D.R. / Tronrud, D.E. / Pley, H.W. / Flaherty, K.M. / Stark, W. / Jansonius, J.N. / McKay, D.B. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1u4g.cif.gz | 77.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1u4g.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1u4g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/u4/1u4gftp://data.pdbj.org/pub/pdb/validation_reports/u4/1u4g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ezmS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 33175.531 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: protein from Nagase, Japan; isolated as extracellular protease from cultures of Pseudomonas aeruginosa Source: (natural) Pseudomonas aeruginosa (bacteria) / Secretion: extracellular / References: UniProt: P14756, pseudolysin |
|---|
-Non-polymers , 5 types, 231 molecules 


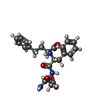

| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
| #3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #5: Chemical | ChemComp-HPI /  Mass: 441.477 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H27N3O6 Mass: 441.477 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H27N3O6 |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 227 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.4 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: ammonium sulfate 1 mM, inhibitor, pH 8, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL11-1 / Wavelength: 0.965 Å / Beamline: BL11-1 / Wavelength: 0.965 Å |
| Detector | Type: ADSC / Detector: CCD |
| Radiation | Monochromator: ssrl bl 11-1 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.965 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→30 Å / Num. obs: 57704 / % possible obs: 89.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rsym value: 0.07 |
| Reflection shell | Resolution: 1.4→1.42 Å / Rsym value: 0.121 / % possible all: 73.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1EZM Resolution: 1.4→30 Å / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 11.82 Å2 | ||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.15 Å / Luzzati d res low obs: 1.4 Å / Luzzati sigma a obs: 0.16 Å | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
