 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1swg | ||||||
|---|---|---|---|---|---|---|---|
| Title | CIRCULAR PERMUTED STREPTAVIDIN E51/A46 IN COMPLEX WITH BIOTIN | ||||||
 Components Components | CIRCULARLY PERMUTED CORE-STREPTAVIDIN E51/A46 | ||||||
 Keywords Keywords | BIOTIN-BINDING PROTEIN / BIOTIN BINDING PROTEIN / CIRCULAR PERMUTATION / BIOTIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Streptomyces avidinii (bacteria) Streptomyces avidinii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Freitag, S. / Chu, V. / Le Trong, I. / Stayton, P.S. / Stenkamp, R.E. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1998 Title: Thermodynamic and structural consequences of flexible loop deletion by circular permutation in the streptavidin-biotin system. Authors: Chu, V. / Freitag, S. / Le Trong, I. / Stenkamp, R.E. / Stayton, P.S. #1: Journal: To be PublishedTitle: Thermodynamic Consequences of the Deletion of a Flexible Loop by Circular Permutation in the Streptavidin-Biotin System Authors: Chu, V. / Freitag, S. / Le Trong, I. / Stenkamp, R.E. / Stayton, P.S. #2: Journal: Protein Sci. / Year: 1997Title: Structural Studies of the Streptavidin Binding Loop Authors: Freitag, S. / Le Trong, I. / Klumb, L. / Stayton, P.S. / Stenkamp, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1swg.cif.gz | 106.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1swg.ent.gz | 82 KB | Display | PDB format |
| PDBx/mmJSON format | 1swg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sw/1swgftp://data.pdbj.org/pub/pdb/validation_reports/sw/1swg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1swfC  1swaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 13329.403 Da / Num. of mol.: 4 Mutation: DELETION OF SURFACE LOOP RESIDUES 45 - 50 FROM THE SEQUENCE. THE OLD N- AND C-TERMINI (S139, A13, RESPECTIVELY) ARE CONNECTED INTRODUCING THE FOUR ADDITIONAL RESIDUES GGGS Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces avidinii (bacteria) / Production host: #2: Chemical | ChemComp-BTN / 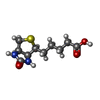  Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S Mass: 244.311 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N2O3S#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 335 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 335 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.87 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.5 Details: PROTEIN (12MG/ML, 10MM BIOTIN) WAS CRYSTALLIZED FROM 52% MPD (PH 4.5) | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 / Beamline: BL9-1 / Wavelength: 0.98 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 1, 1997 / Details: RH-COATED SILICON MIRROR |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→30 Å / Num. obs: 51393 / % possible obs: 98 % / Observed criterion σ(I): -3 / Redundancy: 4 % / Rmerge(I) obs: 0.031 / Net I/σ(I): 19.9 |
| Reflection shell | Resolution: 1.75→1.81 Å / Redundancy: 3 % / Rmerge(I) obs: 0.261 / Mean I/σ(I) obs: 2 / % possible all: 87 |
| Reflection | *PLUS % possible obs: 98.2 % / Num. measured all: 417564 / Rmerge(I) obs: 0.03 |
| Reflection shell | *PLUS % possible obs: 96.2 % / Rmerge(I) obs: 0.17 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SWA (EXCLUDING RESIDUES 46 - 51 IN ALL SUBUNITS) Resolution: 1.8→10 Å / Num. parameters: 15527 / Num. restraintsaints: 17457 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 3354 / Occupancy sum non hydrogen: 3790 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.231 / Rfactor Rwork: 0.181 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 27 Å2 |
