 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qre: A CLOSER LOOK AT THE ACTIVE SITE OF GAMMA-CARBONIC ANHYDRASES: HI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qre | ||||||
|---|---|---|---|---|---|---|---|
| Title | A CLOSER LOOK AT THE ACTIVE SITE OF GAMMA-CARBONIC ANHYDRASES: HIGH RESOLUTION CRYSTALLOGRAPHIC STUDIES OF THE CARBONIC ANHYDRASE FROM METHANOSARCINA THERMOPHILA | ||||||
 Components Components | CARBONIC ANHYDRASE | ||||||
 Keywords Keywords | LYASE / BETA-HELIX | ||||||
| Function / homology |  Function and homology information Function and homology informationbicarbonate binding / sulfate binding / cobalt ion binding / carbonic anhydrase / carbonate dehydratase activity / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Methanosarcina thermophila (archaea) Methanosarcina thermophila (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / ISOMORPHOUS REPLACEMENT / Resolution: 1.46 Å X-RAY DIFFRACTION / SYNCHROTRON / ISOMORPHOUS REPLACEMENT / Resolution: 1.46 Å | ||||||
 Authors Authors | Iverson, T.M. / Alber, B.E. / Kisker, C. / Ferry, J.G. / Rees, D.C. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2000 Title: A closer look at the active site of gamma-class carbonic anhydrases: high-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Authors: Iverson, T.M. / Alber, B.E. / Kisker, C. / Ferry, J.G. / Rees, D.C. #1: Journal: Embo J. / Year: 1996Title: A Left-Handed Beta-Helix revealed by the Crystal Structure of a Carbonic Anhydrase from the Archaeon Methanosarcina thermophila Authors: Kisker, C. / Schindelin, H. / Alber, B. / Ferry, J. / Rees, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qre.cif.gz | 56.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qre.ent.gz | 40 KB | Display | PDB format |
| PDBx/mmJSON format | 1qre.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qr/1qreftp://data.pdbj.org/pub/pdb/validation_reports/qr/1qre | HTTPS FTP |
|---|
-Related structure data
















-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26429.412 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methanosarcina thermophila (archaea) / Production host:  |
|---|---|
| #2: Chemical | ChemComp-BCT / 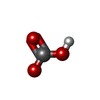  Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM Mass: 61.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM |
| #3: Chemical | ChemComp-CO /   Mass: 58.933 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Co |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.78 Å3/Da / Density % sol: 30.73 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: PEG 8000, amonium sulfate, bicarbonate, pH 5.8, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 6.2 | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 / Beamline: BL7-1 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 11, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.46→20 Å / Num. all: 32929 / Num. obs: 32731 / % possible obs: 99.4 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 3 / Redundancy: 3.9 % / Biso Wilson estimate: 18.7 Å2 / Rmerge(I) obs: 0.055 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 1.46→1.51 Å / Redundancy: 3 % / Rmerge(I) obs: 0.297 / % possible all: 99.1 |
| Reflection | *PLUS Num. measured all: 128698 |
| Reflection shell | *PLUS % possible obs: 99.1 % / Mean I/σ(I) obs: 3.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: ISOMORPHOUS REPLACEMENT / Resolution: 1.46→20 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: refmac
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.46→20 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
