 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qnu: Shiga-Like Toxin I B Subunit Complexed with the Bridged-Starfish ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qnu | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Shiga-Like Toxin I B Subunit Complexed with the Bridged-Starfish Inhibitor | |||||||||
 Components Components | Shiga toxin 1 variant B subunit | |||||||||
 Keywords Keywords | TOXIN / SUBNANOMOLAR INHIBITOR / MULTIVALENT PROTEIN-CARBOHYDRATE RECOGNITION / OB-FOLD | |||||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated modulation of host virulence / symbiont-mediated hemolysis of host erythrocyte / toxin activity / extracellular region Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.23 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.23 Å | |||||||||
 Authors Authors | Pannu, N.S. / Hayakawa, K. / Read, R.J. | |||||||||
 Citation Citation | Journal: Nature / Year: 2000 Title: Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Authors: Kitov, P.I. / Sadowska, J.M. / Mulvey, G. / Armstrong, G.D. / Ling, H. / Pannu, N.S. / Read, R.J. / Bundle, D.R. #1: Journal: Biochemistry / Year: 1998Title: Structure of the Shiga-Like Toxin I B-Pentamer Complexed with an Analogue of its Receptor Bg3 Authors: Ling, H. / Boodhoo, A. / Hazes, B. / Cummings, M.D. / Armstrong, G.D. / Brunton, J.L. / Read, R.J. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qnu.cif.gz | 88 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qnu.ent.gz | 66.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1qnu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qn/1qnuftp://data.pdbj.org/pub/pdb/validation_reports/qn/1qnu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bosS S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||||||
| Details | BIOLOGICAL_UNIT: PENTAMER |
-Components
| #1: Protein | Mass: 7698.634 Da / Num. of mol.: 5 / Fragment: RECEPTOR-BINDING DOMAIN Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH BRIDGE-STARFISH MOLECULE, A SUBNANOMOLAR TAILORED MULTIVALENT INHIBITOR Source: (gene. exp.) #2: Polysaccharide | beta-D-galactopyranose-(1-4)-beta-D-galactopyranose-(1-4)-alpha-D-glucopyranose Source method: isolated from a genetically manipulated source #3: Chemical | ChemComp-EMB / 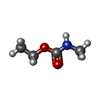  Mass: 103.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H9NO2 Mass: 103.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H9NO2#4: Chemical | ChemComp-MEC / 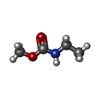  Mass: 103.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H9NO2 Mass: 103.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H9NO2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 80 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 80 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 49 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7 Details: COMPLEX PREPARED BY ADDING 15 MICROLITRES OF BRIDGE-STARFIS (0.35MM) SLOWLY TO 15 MICROLITRES OF SLT-I B-SUBUNIT (10 MG WHILE AGITATING. HANGING DROPS WERE PREPARED BY MIXING THI SOLUTION ...Details: COMPLEX PREPARED BY ADDING 15 MICROLITRES OF BRIDGE-STARFIS (0.35MM) SLOWLY TO 15 MICROLITRES OF SLT-I B-SUBUNIT (10 MG WHILE AGITATING. HANGING DROPS WERE PREPARED BY MIXING THI SOLUTION WITH AN EQUAL VOLUME OF RESERVOIR SOLUTION (28% SA NH4SO4, 2% 2-METHYL-2,4-PENTANEDIOL, 0.1M NACL, 0.1 M HEPES, pH 7.00 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU/MSC RU- / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Feb 15, 1999 / Details: YALE MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.23→50 Å / Num. obs: 19159 / % possible obs: 99 % / Observed criterion σ(I): 0 / Redundancy: 5.3 % / Biso Wilson estimate: 12.8 Å2 / Rmerge(I) obs: 0.148 / Rsym value: 0.148 / Net I/σ(I): 3.4 |
| Reflection shell | Resolution: 2.23→2.35 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.291 / Mean I/σ(I) obs: 2.1 / Rsym value: 0.291 / % possible all: 96.2 |
| Reflection | *PLUS Rmerge(I) obs: 0.161 |
| Reflection shell | *PLUS Lowest resolution: 2.34 Å / % possible obs: 94.9 % / Rmerge(I) obs: 0.339 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1BOS Resolution: 2.23→50 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1625101.21 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 51.4401 Å2 / ksol: 0.411496 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.23→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Rms dev Biso : 4.81 Å2 / Rms dev position: 0.25 Å / Weight Biso : 1 / Weight position: 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.23→2.37 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
