 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ql8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | FACTOR XA SPECIFIC INHIBITOR IN COMPLEX WITH BOVINE TRYPSIN | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | SERINE PROTEASE / HYDROLASE / SERINE PROTEINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 3 Å X-RAY DIFFRACTION / OTHER / Resolution: 3 Å | ||||||
 Authors Authors | Stubbs, M.T. | ||||||
 Citation Citation | Journal: Chembiochem / Year: 2002 Title: Ph-Dependent Binding Modes Observed in Trypsin Crystals: Lessons for the Structure-Based Drug Design Authors: Stubbs, M.T. / Reyda, S. / Dullweber, F. / Moeller, M. / Klebe, G. / Dorsch, D. / Mederski, W.W.K.R. / Wurziger, H. #1: Journal: J.Med.Chem. / Year: 1998 Title: Structural and Functional Analyses of Benzamidine-Based Inhibitors in Complex with Trypsin: Implications for the Inhibition of Factor Xa, Tpa, and Urokinase Authors: Renatus, M. / Bode, W. / Huber, R. / Stuerzebecher, J. / Stubbs, M.T. #2: Journal: Curr.Pharm.Des. / Year: 1996Title: Structural Aspects of Factor Xa Inhibition Authors: Stubbs, M.T. #3: Journal: FEBS Lett. / Year: 1995Title: Crystal Structures of Factor Xa Specific Inhibitors in Complex with Trypsin: Structural Grounds for Inhibition of Factor Xa and Selectivity Against Thrombin Authors: Stubbs, M.T. / Huber, R. / Bode, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ql8.cif.gz | 56.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ql8.ent.gz | 40.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ql8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ql/1ql8ftp://data.pdbj.org/pub/pdb/validation_reports/ql/1ql8 | HTTPS FTP |
|---|
-Related structure data


























-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23324.287 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Chemical | ChemComp-ZEN / [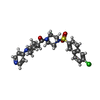  Mass: 499.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27ClN4O3S Mass: 499.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27ClN4O3S |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THE 223 AMINO ACIDS OF BOVINE TRYPSIN ARE IDENTIFIED BY THE RESIDUE NUMBERS OF THE HOMOLOGOUS ...THE 223 AMINO ACIDS OF BOVINE TRYPSIN ARE IDENTIFIED |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.56 Å3/Da / Density % sol: 65.42 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: 0.1M IMIDAZOLE PH7.0, 0.3M AMMONIUM SULPHATE, 30% PEG8K, pH 7.00 | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion / pH: 7 | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 |
| Detector | Type: R-AXIS IV / Detector: IMAGE PLATE / Date: Jul 15, 1998 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→100 Å / Num. obs: 7494 / % possible obs: 99.9 % / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Rmerge(I) obs: 0.147 / Rsym value: 0.147 / Net I/σ(I): 6.4 |
| Reflection shell | Resolution: 2.9→3 Å / Rmerge(I) obs: 0.543 / Rsym value: 0.543 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 3→10 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 10 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3→3.05 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARHCSDX.PRO / Topol file: TOPHCSDX.PRO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.19 / Rfactor Rwork: 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.27 |
