 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1q4v: CRYSTAL STRUCTURE OF ALLO-ILEA2-INSULIN, AN INACTIVE CHIRAL ANALO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1q4v | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF ALLO-ILEA2-INSULIN, AN INACTIVE CHIRAL ANALOGUE: IMPLICATIONS FOR THE MECHANISM OF RECEPTOR | |||||||||
 Components Components | (Insulin) x 2 | |||||||||
 Keywords Keywords | HORMONE/GROWTH FACTOR / allo-Ile-A2-insulin / protein unfolding / insulin receptor / HORMONE-GROWTH FACTOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of glycogen catabolic process / : / negative regulation of fatty acid metabolic process / Signaling by Insulin receptor / IRS activation / Insulin processing / regulation of protein secretion / positive regulation of peptide hormone secretion / negative regulation of feeding behavior / negative regulation of acute inflammatory response ...negative regulation of glycogen catabolic process / : / negative regulation of fatty acid metabolic process / Signaling by Insulin receptor / IRS activation / Insulin processing / regulation of protein secretion / positive regulation of peptide hormone secretion / negative regulation of feeding behavior / negative regulation of acute inflammatory response / Regulation of gene expression in beta cells / positive regulation of respiratory burst / alpha-beta T cell activation / Synthesis, secretion, and deacylation of Ghrelin / negative regulation of protein secretion / positive regulation of dendritic spine maintenance / negative regulation of gluconeogenesis / fatty acid homeostasis / positive regulation of glycogen biosynthetic process / positive regulation of insulin receptor signaling pathway / Signal attenuation / FOXO-mediated transcription of oxidative stress, metabolic and neuronal genes / negative regulation of lipid catabolic process / negative regulation of respiratory burst involved in inflammatory response / positive regulation of lipid biosynthetic process / negative regulation of oxidative stress-induced intrinsic apoptotic signaling pathway / nitric oxide-cGMP-mediated signaling / regulation of protein localization to plasma membrane / positive regulation of nitric-oxide synthase activity / transport vesicle / Insulin receptor recycling / COPI-mediated anterograde transport / negative regulation of reactive oxygen species biosynthetic process / positive regulation of brown fat cell differentiation / insulin-like growth factor receptor binding / NPAS4 regulates expression of target genes / neuron projection maintenance / positive regulation of mitotic nuclear division / endoplasmic reticulum-Golgi intermediate compartment membrane / Insulin receptor signalling cascade / positive regulation of glycolytic process / endosome lumen / acute-phase response / positive regulation of cytokine production / positive regulation of D-glucose import across plasma membrane / insulin receptor binding / positive regulation of long-term synaptic potentiation / positive regulation of protein secretion / positive regulation of cell differentiation / wound healing / Regulation of insulin secretion / positive regulation of neuron projection development / hormone activity / negative regulation of protein catabolic process / positive regulation of protein localization to nucleus / regulation of synaptic plasticity / glucose metabolic process / Golgi lumen / cognition / vasodilation / insulin receptor signaling pathway / glucose homeostasis / cell-cell signaling / regulation of protein localization / PI5P, PP2A and IER3 Regulate PI3K/AKT Signaling / positive regulation of cell growth / protease binding / secretory granule lumen / positive regulation of MAPK cascade / positive regulation of canonical NF-kappaB signal transduction / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / positive regulation of cell migration / endoplasmic reticulum lumen / G protein-coupled receptor signaling pathway / Amyloid fiber formation / receptor ligand activity / Golgi membrane / negative regulation of gene expression / positive regulation of cell population proliferation / positive regulation of gene expression / regulation of DNA-templated transcription / : / extracellular region / identical protein binding Similarity search - Function | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | |||||||||
 Authors Authors | Wan, Z.L. / Xu, B. / Chu, Y.C. / Katsoyannis, P.G. / Weiss, M.A. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Crystal structure of allo-Ile(A2)-insulin, an inactive chiral analogue: implications for the mechanism of receptor binding. Authors: Wan, Z.L. / Xu, B. / Chu, Y.C. / Katsoyannis, P.G. / Weiss, M.A. #1: Journal: J.Mol.Biol. / Year: 2002Title: CHIRAL MUTAGENESIS OF INSULIN'S HIDDEN RECEPTOR-BINDING SURFACE: STRUCTURE OF AN ALLO-ISOLEUCINE (A2) ANALOGUE Authors: Xu, B. / Hua, Q.X. / NAKAGAWA, S.H. / JIA, W. / CHU, Y.C. / KASOYANNIS, P.G. / WEISS, M.A. #2: Journal: Protein Sci. / Year: 2002Title: A CAVITY-FORMING MUTATION IN INSULIN INDUCES SEGMENTAL UNFOLDING OF A SURROUNDING ALPHA-HELIX Authors: XU, B. / HUA, Q.X. / NAKAGAWA, S.H. / JIA, W. / CHU, Y.C. / KATSOYANNIS, P.G. / WEISS, M.A. #3: Journal: J.Mol.Biol. / Year: 2002Title: NON-STANDARD INSULIN DESIGN: STRUCTURE-ACTIVITY RELATIONSHIPS AT THE PERIPHERY OF THE INSULIN Receptor Authors: WEISS, M.A. / WAN, Z. / ZHAO, M. / CHU, Y.C. / NAKAGAWA, S.H. / BURKE, G.T. / JIA, W. / HELLMICH, R. / KATSOYANNIS, P.G. #4: Journal: Trends Biochem.Sci. / Year: 1999Title: IS PROTEIN FOLDING HIERARCHIC? I. LOCAL STRUCTURE AND PEPTIDE FOLDING Authors: BALDWIN, R.L. / ROSE, G.D. #5: Journal: Trends Biochem.Sci. / Year: 1999Title: IS PROTEIN FOLDING HIERARCHIC? II. FOLDING INTERMEDIATES AND TRANSITION STATES Authors: BALDWIN, R.L. / ROSE, G.D. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1q4v.cif.gz | 35.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1q4v.ent.gz | 24.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1q4v.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q4/1q4vftp://data.pdbj.org/pub/pdb/validation_reports/q4/1q4v | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1trzS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

















- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| 3 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | The crystallographic asymmetry unit of insulin consists of two monomers each consisting two heterochains. The entry presents coordinates for monomer 1 (chain indicators A and B)and monomer 2 (chain indicators C and D). There are two zinc ions per insulin hexamer located on the three-fold axis. The conformations of two monomers are different the result of B changed in conformation of the first residues of the B-chain. The biological assembly is a hexamer generated from the dimer in the asymmetric unit by the operations: -y,x-y,z and -x+y,-x,z |
-Components
| #1: Protein/peptide | Mass: 2383.698 Da / Num. of mol.: 2 / Fragment: INSULIN A CHAIN / Source method: obtained synthetically Details: THE PROTEIN WAS CHEMICALLY SYNTHESIZED. THE SEQUENCE OF THE PROTEIN IS NATURALLY FOUND IN Homo Sapiens (human). References: GenBank: AAA59172, UniProt: P01308*PLUS #2: Protein/peptide | Mass: 3433.953 Da / Num. of mol.: 2 / Fragment: INSULIN B CHAIN / Source method: obtained synthetically Details: THE PROTEIN WAS CHEMICALLY SYNTHESIZED. THE SEQUENCE OF THE PROTEIN IS NATURALLY FOUND IN Homo Sapiens (human). References: GenBank: AAA59172, UniProt: P01308*PLUS #3: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-IPH / | 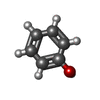  Mass: 94.111 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6O Mass: 94.111 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6O#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.48 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 6.4 Details: Tris, sodium citrate, acetone, phenol, pH 6.4, VAPOR DIFFUSION, HANGING DROP, temperature 290.0K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Nov 12, 2001 / Details: mirrors |
| Radiation | Monochromator: mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→23.24 Å / Num. obs: 5897 / % possible obs: 98.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.07 % / Biso Wilson estimate: 26.2 Å2 / Rmerge(I) obs: 0.057 / Net I/σ(I): 43.1 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.301 / Mean I/σ(I) obs: 4.1 / Num. unique all: 543 / % possible all: 89.3 |
| Reflection | *PLUS Lowest resolution: 23.2 Å / Num. obs: 5892 |
| Reflection shell | *PLUS % possible obs: 89.3 % / Redundancy: 1.27 % / Num. unique obs: 543 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1TRZ Resolution: 2→23.24 Å / Rfactor Rfree error: 0.006 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: Flat model / Bsol: 37.6603 Å2 / ksol: 0.29202 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→23.24 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.011 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 23.2 Å | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.07 Å |
