 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1psa: STRUCTURE OF A PEPSIN(SLASH)RENIN INHIBITOR COMPLEX REVEALS A NOV... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1psa | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF A PEPSIN(SLASH)RENIN INHIBITOR COMPLEX REVEALS A NOVEL CRYSTAL PACKING INDUCED BY MINOR CHEMICAL ALTERATIONS IN THE INHIBITOR | ||||||
 Components Components | PEPSIN A | ||||||
 Keywords Keywords | HYDROLASE/hydrolase inhibitor / ACID PROTEINASE / HYDROLASE-hydrolase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationSurfactant metabolism / pepsin A / digestion / aspartic-type endopeptidase activity / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.9 Å X-RAY DIFFRACTION / Resolution: 2.9 Å | ||||||
 Authors Authors | Chen, L. / Abad-Zapatero, C. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.B / Year: 1992 Title: Structure of a pepsin/renin inhibitor complex reveals a novel crystal packing induced by minor chemical alterations in the inhibitor. Authors: Chen, L. / Erickson, J.W. / Rydel, T.J. / Park, C.H. / Neidhart, D. / Luly, J. / Abad-Zapatero, C. #1: Journal: Adv.Exp.Med.Biol. / Year: 1991Title: Inhibitor Binding Induces Structural Changes in Porcine Pepsin Authors: Abad-Zapatero, C. / Rydel, T.J. / Neidhart, D.J. / Luly, J. / Erickson, J.W. #2: Journal: Proteins / Year: 1990Title: Revised 2.3 Angstroms Structure of Porcine Pepsin: Evidence for a Flexible Subdomain Authors: Abad-Zapatero, C. / Rydel, T.J. / Erickson, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1psa.cif.gz | 134.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1psa.ent.gz | 104.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1psa.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ps/1psaftp://data.pdbj.org/pub/pdb/validation_reports/ps/1psa | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES PRO A 23 AND PRO B 23 ARE CIS PROLINES. 2: RESIDUES 239 - 242 AND 278 - 281 OF CHAINS *A* AND *B* HAD THE WEAKEST ELECTRON DENSITY. | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.786, 0.036, 0.617), Vector: Details | THE TRANSFORMATION PRESENTED ON *MTRIX* RECORDS BELOW WILL YIELD APPROXIMATE COORDINATES FOR CHAIN *B* WHEN APPLIED TO CHAIN *A*. | |
-Components
| #1: Protein | Mass: 34513.508 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 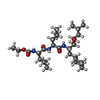  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 541.763 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C29H55N3O6 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 541.763 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C29H55N3O6References: N-(ethoxycarbonyl)-L-leucyl-N-[(1R,2S,3S)-1-(cyclohexylmethyl)-2,3-dihydroxy-5-methylhexyl]-L-leucinamide #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 110 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 110 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | THE INHIBITOR A-62095 IS A TETRAPEPTIDE ANALOGUE WITH A T-BOC BLOCKING GROUP AND CONTAINING ...THE INHIBITOR A-62095 IS A TETRAPEPTI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 42.98 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 2 / Method: unknownDetails: taken from Andreeva, N.S. et al (1984). J. Biol. Chem., 259, 11353-11365. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.9 Å / Lowest resolution: 3 Å / Num. all: 13736 / Num. obs: 11951 / % possible obs: 87 % / Num. measured all: 24379 / Rmerge(I) obs: 0.044 |
- Processing
Processing
| Software | Name: X-PLOR / Classification: refinement | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.9→8 Å / Rfactor Rwork: 0.139 Details: THE SPACE GROUP OF THIS STRUCTURE IS MONOCLINIC P21, WITH C AXIS UNIQUE. THE ASYMMETRIC UNIT CONTAINS TWO PEPSIN/ INHIBITOR COMPLEXES. RESIDUES 239 - 242 AND 278 - 281 OF CHAINS *A* AND *B* ...Details: THE SPACE GROUP OF THIS STRUCTURE IS MONOCLINIC P21, WITH C AXIS UNIQUE. THE ASYMMETRIC UNIT CONTAINS TWO PEPSIN/ INHIBITOR COMPLEXES. RESIDUES 239 - 242 AND 278 - 281 OF CHAINS *A* AND *B* HAD THE WEAKEST ELECTRON DENSITY. | ||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→8 Å
| ||||||||||||
| Refine LS restraints |
| ||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||
| Refinement | *PLUS Highest resolution: 2.9 Å / Lowest resolution: 8 Å / Num. reflection all: 11284 / Rfactor obs: 0.139 | ||||||||||||
| Solvent computation | *PLUS | ||||||||||||
| Displacement parameters | *PLUS | ||||||||||||
| Refine LS restraints | *PLUS
|
