 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pob: CRYSTAL STRUCTURE OF COBRA-VENOM PHOSPHOLIPASE A2 IN A COMPLEX WI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pob | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF COBRA-VENOM PHOSPHOLIPASE A2 IN A COMPLEX WITH A TRANSITION-STATE ANALOGUE | ||||||
 Components Components | PHOSPHOLIPASE A2 | ||||||
 Keywords Keywords | HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationphospholipase A2 / : / arachidonate secretion / lipid catabolic process / phospholipid metabolic process / phospholipid binding / positive regulation of fibroblast proliferation / fatty acid biosynthetic process / toxin activity / signaling receptor binding ...phospholipase A2 / : / arachidonate secretion / lipid catabolic process / phospholipid metabolic process / phospholipid binding / positive regulation of fibroblast proliferation / fatty acid biosynthetic process / toxin activity / signaling receptor binding / calcium ion binding / extracellular region Similarity search - Function | ||||||
| Biological species |  Naja atra (Chinese cobra) Naja atra (Chinese cobra) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2 Å X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | White, S.P. / Scott, D.L. / Otwinowski, Z. / Sigler, P.B. | ||||||
 Citation Citation | Journal: Science / Year: 1990 Title: Crystal structure of cobra-venom phospholipase A2 in a complex with a transition-state analogue. Authors: White, S.P. / Scott, D.L. / Otwinowski, Z. / Gelb, M.H. / Sigler, P.B. #1: Journal: Science / Year: 1990Title: Interfacial Catalysis: The Mechanism of Phospholipase A2 Authors: Scott, D.L. / White, S.P. / Otwinowski, Z. / Yuan, W. / Gelb, M.H. / Sigler, P.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pob.cif.gz | 67.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pob.ent.gz | 47.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1pob.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/po/1pobftp://data.pdbj.org/pub/pdb/validation_reports/po/1pob | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13154.642 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Naja atra (Chinese cobra) / References: UniProt: P00598, phospholipase A2#2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical | 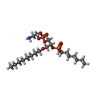  Mass: 489.521 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H45NO8P2 Mass: 489.521 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H45NO8P2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 242 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 242 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.93 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2 Å / Rmerge(I) obs: 0.07 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor Rwork: 0.179 / Rfactor obs: 0.179 / Highest resolution: 2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Rfactor obs: 0.179 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
