 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1po3: Crystal structure of ferric citrate transporter FecA in complex w... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1po3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of ferric citrate transporter FecA in complex with ferric citrate | ||||||
 Components Components | Iron(III) dicitrate transport protein fecA precursor | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / outer membrane protein / beta barrel / TonB-dependent transport / citrate / siderophore / iron | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to iron ion starvation / siderophore-iron transmembrane transporter activity / Metal ion assimilation from the host / siderophore-iron import into cell / siderophore transport / transmembrane transporter complex / signal transduction involved in regulation of gene expression / cell outer membrane / signaling receptor activity / intracellular iron ion homeostasis ...response to iron ion starvation / siderophore-iron transmembrane transporter activity / Metal ion assimilation from the host / siderophore-iron import into cell / siderophore transport / transmembrane transporter complex / signal transduction involved in regulation of gene expression / cell outer membrane / signaling receptor activity / intracellular iron ion homeostasis / regulation of DNA-templated transcription / membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å | ||||||
 Authors Authors | Yue, W.W. / Grizot, S. / Buchanan, S.K. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2003 Title: Structural evidence for iron-free citrate and ferric citrate binding to the TonB-dependent outer membrane transporter FecA Authors: Yue, W.W. / Grizot, S. / Buchanan, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1po3.cif.gz | 243.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1po3.ent.gz | 194.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1po3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/po/1po3ftp://data.pdbj.org/pub/pdb/validation_reports/po/1po3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1pnzC  1po0C  1kmpS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj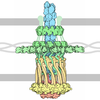






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a monomer although asymmetric unit is an NCS-related dimer |
-Components
| #1: Protein | Mass: 83169.328 Da / Num. of mol.: 2 / Fragment: FecA residues 95-741 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-FLC / 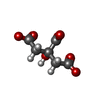  Mass: 189.100 Da / Num. of mol.: 4 / Fragment: diferric dicitrate / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 4 / Fragment: diferric dicitrate / Source method: obtained synthetically / Formula: C6H5O7#3: Chemical | ChemComp-FE /   Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.4 Å3/Da / Density % sol: 63.8 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG1000, LDAO, calcium chloride, Bis-Tris, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 295K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 8 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X9B / Wavelength: 0.9795 Å / Beamline: X9B / Wavelength: 0.9795 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Feb 17, 2002 |
| Radiation | Monochromator: synchrotron / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 3.4→19.77 Å / Num. all: 33661 / Num. obs: 30446 / % possible obs: 99.2 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 4.4 % / Biso Wilson estimate: 78.7 Å2 / Rmerge(I) obs: 0.097 / Net I/σ(I): 13.9 |
| Reflection shell | Resolution: 3.4→3.52 Å / Redundancy: 4.4 % / Rmerge(I) obs: 0.36 / Mean I/σ(I) obs: 3.8 / Num. unique all: 3036 / % possible all: 100 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. obs: 30794 / % possible obs: 100 % / Num. measured all: 135112 |
| Reflection shell | *PLUS % possible obs: 100 % / Num. unique obs: 3036 / Num. measured obs: 13363 / Rmerge(I) obs: 0.382 / Mean I/σ(I) obs: 3.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB accession code 1KMP Resolution: 3.4→15 Å / Rfactor Rfree error: 0.008 / Isotropic thermal model: GROUP / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: In chain A side chains were not built for residues GLN221, MET249, ARG336, GLN351, ARG385, MET396, TYR499, ASP503, GLU533, ARG582, THR600, GLU616, ASP622, TYR624, LYS668, GLN695 and LYS740. ...Details: In chain A side chains were not built for residues GLN221, MET249, ARG336, GLN351, ARG385, MET396, TYR499, ASP503, GLU533, ARG582, THR600, GLU616, ASP622, TYR624, LYS668, GLN695 and LYS740. In chain B side chains were not built for residues ARG217, ASP222, MET249, HIS295, GLU307, PHE346, GLN351, ARG385, MET396, GLN428, TYR436, LYS454, MET466, GLU485, GLU533, ASP548, TYR592, LYS620 and MET696.
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 10 Å2 / ksol: 0.268617 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 57.4 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.4→15 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.4→3.61 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3.4 Å / Lowest resolution: 15 Å / % reflection Rfree: 5 % | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
