 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1p4y | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Effect of Sequence on the Conformational Geometry of DNA Holliday Junctions | ||||||||||||||||||
 要素 要素 | 5'-D(* キーワード キーワードDNA / Holliday Junction / DNA Four-Way Junction | 機能・相同性 | DNA |  機能・相同性情報 機能・相同性情報手法 |  X線回折 / 分子置換 / 解像度: 1.7 Å X線回折 / 分子置換 / 解像度: 1.7 Å  データ登録者 データ登録者Hays, F.A. / Vargason, J.M. / Ho, P.S. |  引用ジャーナル: Biochemistry / 年: 2003 引用ジャーナル: Biochemistry / 年: 2003タイトル: Effect of Sequence on the Conformation of DNA Holliday Junctions 著者: Hays, F.A. / Vargason, J.M. / Ho, P.S. 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1p4y.cif.gz | 21.9 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1p4y.ent.gz | 14.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1p4y.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1p4y_validation.pdf.gz | 383.9 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1p4y_full_validation.pdf.gz | 384.5 KB | 表示 | |
| XML形式データ | 1p4y_validation.xml.gz | 4.1 KB | 表示 | |
| CIF形式データ | 1p4y_validation.cif.gz | 5.6 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/p4/1p4yftp://data.pdbj.org/pub/pdb/validation_reports/p4/1p4y | HTTPS FTP |
-関連構造データ





-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 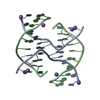
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF TWO CHAINS A AND B. THE FULL FOUR STRANDED HOLLIDAY JUNCTION STRUCTURE CAN BE GENERATED BY THE FOLLOWING: matrix= ( -1.00000 -0.00046 -0.00015 ) ( -0.00046 1.00000 0.00072 ) ( 0.00015 0.00072 -1.00000 ) translation= (-12.65734 -0.02663 34.77174) |
-要素
| #1: DNA鎖 | 分子量: 3046.980 Da / 分子数: 2 / 由来タイプ: 合成 詳細: DNA was synthesized on an Applied Biosystems DNA synthesizer using phosphoramidite chemistry, with the trityl-protecting group left intact at the 5-terminal nucleotide, FOR SUBSEQUENT HPLC ...詳細: DNA was synthesized on an Applied Biosystems DNA synthesizer using phosphoramidite chemistry, with the trityl-protecting group left intact at the 5-terminal nucleotide, FOR SUBSEQUENT HPLC PURIFICATION, THEN DEPROTECTED BY TREATMENT WITH 3% ACETIC ACID FOR FIFTEEN MINUTES, NEUTRALIZED WITH AMMONIUM HYDROXIDE, AND DESALTED ON A SIGMA G-25 SEPHADEX COLUMN. #2: 化合物 | ChemComp-NA /   分子量: 22.990 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Na 分子量: 22.990 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Na#3: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 108 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 108 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 1.94 Å3/Da / 溶媒含有率: 35.95 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 298 K / pH: 7 詳細: 0.5mM DNA, 25mM sodium cacodylate, 7.5mM CaCl2, 0.1mM SPERMINE TETRAHYDROCHLORIDE, pH 7.0, VAPOR DIFFUSION, SITTING DROP, temperature 298K, pH 7.00 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 25 ℃ / pH: 7 / 手法: 蒸気拡散法 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 103 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RU300 / 波長: 1.542 |
| 検出器 | タイプ: RIGAKU RAXIS IV / 検出器: IMAGE PLATE / 日付: 2003年1月24日 |
| 放射 | モノクロメーター: NI FILTER / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.542 Å / 相対比: 1 |
| 反射 | 解像度: 1.65→22.55 Å / Num. obs: 4940 / % possible obs: 74 % / Observed criterion σ(I): 0 / Biso Wilson estimate: 1.6 Å2 / Rmerge(I) obs: 0.048 |
| 反射 シェル | 解像度: 1.65→1.71 Å / Rmerge(I) obs: 0.279 / Mean I/σ(I) obs: 2.14 / % possible all: 21.7 |
| 反射 | *PLUS 最高解像度: 1.6 Å / 最低解像度: 50 Å / Num. obs: 5137 / % possible obs: 78.7 % / Rmerge(I) obs: 0.049 / Num. measured all: 50581 |
| 反射 シェル | *PLUS % possible obs: 48 % / Rmerge(I) obs: 0.278 / Mean I/σ(I) obs: 2.7 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: UD0008 解像度: 1.7→22.5 Å / Rfactor Rfree error: 0.012 / 交差検証法: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 8.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→22.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.7→1.81 Å / Rfactor Rfree error: 0.046 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最高解像度: 1.7 Å / 最低解像度: 20 Å / Rfactor Rfree: 0.26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
