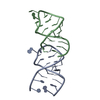
Entry Database : PDB / ID : 1or3Title APOLIPOPROTEIN E3 (APOE3), TRIGONAL TRUNCATION MUTANT 165 PROTEIN (APOLIPOPROTEIN E) Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / Resolution : 1.73 Å Authors Rupp, B. / Segelke, B.W. Journal : Protein Sci. / Year : 2000Title : Conformational flexibility in the apolipoprotein E amino-terminal domain structure determined from three new crystal forms: implications for lipid binding.Authors : Segelke, B.W. / Forstner, M. / Knapp, M. / Trakhanov, S.D. / Parkin, S. / Newhouse, Y.M. / Bellamy, H.D. / Weisgraber, K.H. / Rupp, B. History Deposition Dec 1, 1998 Deposition site / Processing site Revision 1.0 May 24, 2000 Provider / Type Revision 1.1 Apr 26, 2008 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Oct 4, 2017 Group / Category / Item Revision 1.4 Dec 27, 2023 Group / Database references / Category / chem_comp_bond / database_2Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj











 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing