 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nhk: CRYSTAL STRUCTURE OF MYXOCOCCUS XANTHUS NUCLEOSIDE DIPHOSPHATE KI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nhk | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF MYXOCOCCUS XANTHUS NUCLEOSIDE DIPHOSPHATE KINASE AND ITS INTERACTION WITH A NUCLEOTIDE SUBSTRATE AT 2.0 ANGSTROMS RESOLUTION | ||||||
 Components Components | NUCLEOSIDE DIPHOSPHATE KINASE | ||||||
 Keywords Keywords | PHOSPHOTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationnucleoside-diphosphate kinase / UTP biosynthetic process / CTP biosynthetic process / nucleoside diphosphate kinase activity / GTP biosynthetic process / ATP binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Myxococcus xanthus (bacteria) Myxococcus xanthus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Strelkov, S. / Williams, R.L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1995 Title: The 1.9 A crystal structure of a nucleoside diphosphate kinase complex with adenosine 3',5'-cyclic monophosphate: evidence for competitive inhibition. Authors: Strelkov, S.V. / Perisic, O. / Webb, P.A. / Williams, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nhk.cif.gz | 74.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nhk.ent.gz | 56.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1nhk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nh/1nhkftp://data.pdbj.org/pub/pdb/validation_reports/nh/1nhk | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.0008, -0.9883, 0.1525), Vector: Details | MTRIX THE TRANSFORMATIONS PRESENTED ON MTRIX RECORDS BELOW DESCRIBE NON-CRYSTALLOGRAPHIC RELATIONSHIPS AMONG THE VARIOUS DOMAINS IN THIS ENTRY. APPLYING THE APPROPRIATE MTRIX TRANSFORMATION TO THE RESIDUES LISTED FIRST WILL YIELD APPROXIMATE COORDINATES FOR THE RESIDUES LISTED SECOND. APPLIED TO TRANSFORMED TO MTRIX RESIDUES RESIDUES RMSD M1 L 2 - L 144 R 2 - R 144 0.230 | |
-Components
| #1: Protein | Mass: 16025.375 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Myxococcus xanthus (bacteria) / Gene: CMP / Plasmid: PUC119 DERIVED / Production host: #2: Chemical | 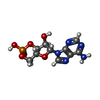  Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 219 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 219 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | FOR EACH POLYPEPTIDE CHAIN THE SINGLE CAMP MOLECULE BOUND TO IT WAS MODELED WITH TWO ALTERNATIVE ...FOR EACH POLYPEPTID | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.73 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.7 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Num. obs: 26296 / % possible obs: 99.1 % / Observed criterion σ(I): 0 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 27 Å / Redundancy: 4.3 % / Num. measured all: 113713 / Rmerge(I) obs: 0.052 |
| Reflection shell | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 2 Å / Rmerge(I) obs: 0.206 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→6 Å / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_dihedral_angle_deg / Dev ideal: 22.4 |
