 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nf7: Ternary complex of the human type II Inosine Monophosphate Dedhyd... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nf7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Ternary complex of the human type II Inosine Monophosphate Dedhydrogenase with Ribavirin Monophosphate and C2-Mycophenolic Adenine Dinucleotide | |||||||||
 Components Components | Inosine-5'-monophosphate dehydrogenase 2 | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / 8 STRANDED PARALLE ALPHA/BETA BARREL / DEHYDROGENASE / IMPD / IMPDH / RIBAVIRIN MONOPHOSPHATE / C2-MAD | |||||||||
| Function / homology |  Function and homology information Function and homology informationlymphocyte proliferation / 'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / Azathioprine ADME / peroxisomal membrane / GTP biosynthetic process / cellular response to interleukin-4 ...lymphocyte proliferation / 'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / Azathioprine ADME / peroxisomal membrane / GTP biosynthetic process / cellular response to interleukin-4 / circadian rhythm / secretory granule lumen / Potential therapeutics for SARS / ficolin-1-rich granule lumen / nucleotide binding / Neutrophil degranulation / DNA binding / RNA binding / extracellular exosome / extracellular region / membrane / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å | |||||||||
 Authors Authors | Risal, D. / Strickler, M.D. / Goldstein, B.M. | |||||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of Human Inosine Monophosphate Dehydrogenase type II complexed with the MPA/NAD analog C2-MAD Authors: Risal, D. / Strickler, M.D. / Goldstein, B.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nf7.cif.gz | 187.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nf7.ent.gz | 148.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1nf7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nf/1nf7ftp://data.pdbj.org/pub/pdb/validation_reports/nf/1nf7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b3oS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 55874.863 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: IMPDH2 / Plasmid: pET12B / Species (production host): Escherichia coli / Production host:  |
|---|
-Non-polymers , 5 types, 110 molecules 
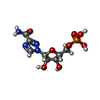
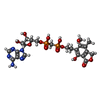

| #2: Chemical |  Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#3: Chemical |  Mass: 324.185 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H13N4O8P Mass: 324.185 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H13N4O8P#4: Chemical |  Mass: 645.450 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H29N5O13P2 Mass: 645.450 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H29N5O13P2#5: Chemical | ChemComp-UNL / Mass: 103.120 Da / Num. of mol.: 11 / Source method: obtained synthetically #6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.48 Å3/Da / Density % sol: 64.34 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 6% PEG 6000, 0.1M Tris-HCl, 24mM beta-mercaptoethanol, 1M LiCl, 10% Glycerol, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 277.0K |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.93 Å / Beamline: A1 / Wavelength: 0.93 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Mar 3, 2002 / Details: mirrors |
| Radiation | Monochromator: Silicon / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.93 Å / Relative weight: 1 |
| Reflection | Resolution: 2.65→41.24 Å / Num. all: 39557 / Num. obs: 34951 / % possible obs: 89 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.5 % / Biso Wilson estimate: 30.7 Å2 / Rmerge(I) obs: 0.038 / Rsym value: 0.038 / Net I/σ(I): 16 |
| Reflection shell | Resolution: 2.65→2.72 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.371 / Mean I/σ(I) obs: 2 / Num. unique all: 2383 / Rsym value: 0.371 / % possible all: 89 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Chain A of PDB ENTRY 1B3O Resolution: 2.65→41.24 Å / Rfactor Rfree error: 0.006 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1.1 / Stereochemistry target values: Engh & Huber Details: BULK SOLVENT MODEL USED. The residues labelled UNK 801 through 811 represent residues whose electron density is robust enough only to trace the main chain. It is not clear how these atoms ...Details: BULK SOLVENT MODEL USED. The residues labelled UNK 801 through 811 represent residues whose electron density is robust enough only to trace the main chain. It is not clear how these atoms correspond to the sequence assignment. The CA atoms are intended to communicate the fact that there is significant density (visible at 0.6 sigma or lower) in the region of the active site that is very near to the bound cofactor analog and therefore potentially making interactions with the analog.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 34.1117 Å2 / ksol: 0.339281 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.7 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→41.24 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.65→2.82 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
