 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1na5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | INTEGRIN ALPHA M I DOMAIN | ||||||
 Components Components | Integrin alpha-M | ||||||
 Keywords Keywords | CELL ADHESION / Rossmann fold | ||||||
| Function / homology |  Function and homology information Function and homology informationectodermal cell differentiation / integrin alphaM-beta2 complex / positive regulation of neutrophil degranulation / response to Gram-positive bacterium / response to curcumin / leukocyte adhesion to vascular endothelial cell / positive regulation of microglial cell mediated cytotoxicity / vertebrate eye-specific patterning / complement component C3b binding / complement-mediated synapse pruning ...ectodermal cell differentiation / integrin alphaM-beta2 complex / positive regulation of neutrophil degranulation / response to Gram-positive bacterium / response to curcumin / leukocyte adhesion to vascular endothelial cell / positive regulation of microglial cell mediated cytotoxicity / vertebrate eye-specific patterning / complement component C3b binding / complement-mediated synapse pruning / Toll Like Receptor 4 (TLR4) Cascade / cell-cell adhesion mediated by integrin / complement receptor mediated signaling pathway / integrin complex / heterotypic cell-cell adhesion / cargo receptor activity / phagocytosis, engulfment / forebrain development / negative regulation of dopamine metabolic process / amyloid-beta clearance / tertiary granule membrane / plasma membrane raft / Integrin cell surface interactions / positive regulation of protein targeting to membrane / response to mechanical stimulus / specific granule membrane / positive regulation of superoxide anion generation / heat shock protein binding / receptor-mediated endocytosis / response to ischemia / response to amphetamine / integrin-mediated signaling pathway / cell-matrix adhesion / Cell surface interactions at the vascular wall / cell-cell adhesion / microglial cell activation / integrin binding / response to estradiol / amyloid-beta binding / signaling receptor activity / Interleukin-4 and Interleukin-13 signaling / cell adhesion / external side of plasma membrane / innate immune response / Neutrophil degranulation / cell surface / : / extracellular exosome / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | McCleverty, C.J. / Liddington, R.C. | ||||||
 Citation Citation | Journal: Biochem.J. / Year: 2003 Title: Engineered allosteric mutants of the integrin alphaMbeta2 I domain: structural and functional studies Authors: McCleverty, C.J. / Liddington, R.C. | ||||||
| History |
|
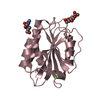
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1na5.cif.gz | 56.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1na5.ent.gz | 39.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1na5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/na/1na5ftp://data.pdbj.org/pub/pdb/validation_reports/na/1na5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1mf7C  1n9zC  1jlmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22461.680 Da / Num. of mol.: 1 / Fragment: Alpha M I domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ITGAM OR CR3A OR CD11B / Plasmid: pGEX-2T / Production host:  |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.05 Å3/Da / Density % sol: 39.38 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 4.2 Details: PEG 8000, sodium chloride, potassium citrate, pH 4.2, VAPOR DIFFUSION, SITTING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 23 ℃ / pH: 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.976 Å / Beamline: BL9-1 / Wavelength: 0.976 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: May 2, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→20 Å / Num. all: 35988 / Num. obs: 31466 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Biso Wilson estimate: 15.6 Å2 |
| Reflection shell | Resolution: 1.5→1.56 Å / % possible all: 99.3 |
| Reflection | *PLUS Highest resolution: 1.5 Å / Num. obs: 35988 / Num. measured all: 125137 / Rmerge(I) obs: 0.025 |
| Reflection shell | *PLUS % possible obs: 92.9 % / Rmerge(I) obs: 0.222 / Mean I/σ(I) obs: 3.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1JLM Resolution: 1.5→20 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 713495.02 / Data cutoff high rms absF: 713495.02 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 73.7649 Å2 / ksol: 0.446952 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.7 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→20 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.59 Å / Rfactor Rfree error: 0.016 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.5 Å / Lowest resolution: 20 Å / Num. reflection obs: 34414 / % reflection Rfree: 5 % / Rfactor Rfree: 0.227 / Rfactor Rwork: 0.205 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
